Richard A. Miller is a Professor of Pathology at the University of Michigan and the Director of Michigan’s Paul F. Glenn Center for Biology of Aging Research. He is also a driving force behind the ITP, the Interventions Testing Program, created in the early 2000s to study the effect of various drugs on lifespan in mice. The ITP is a unique undertaking, with drugs being tested simultaneously in three research facilities to achieve “instant reproducibility” and increased statistical power. The ITP has produced evidence for anti-aging drugs, such as rapamycin, and, in contrast, has shown a lack of effectiveness for several agents, such as metformin. We discuss the ITP findings, including some that have not been published yet, and other geroscience-related topics.
You once said about your early interest in aging: “I decided early on that aging was bad for you. It made people sick and then die”. This sounds so simple and true. Why do you think many people still don’t take seriously the idea that aging can and should be tackled?
People are easily cowed by scientific information. They get a lot of it, and much of the information comes from people who think about aging in ways that appeal to fantasy and to wish fulfillment. Public personae who talk about aging usually are making things up and hyping it without a lot of detailed evidence behind what they’re saying.
It makes smart people skeptical, and it’s harder for people who actually have some information to rise above that in terms of clarity. If you do a Google search for anti-aging medicines, most of what you get is stuff that somebody wants to sell you and make a profit on but which doesn’t actually work. There’s just no evidence that it can work in people or even in mice. With that kind of a noisy environment, it’s hard for the more accurate information to come to public attention.
So, it’s long been a sort of a truism (not true in this case), that nothing can be done about aging. Once you assume that all is lost, everyone’s going to get old, et cetera, then counterclaims become less credible. No one, of course, is claiming that aging can be prevented completely. That would be fantasy. But there’s now very strong evidence, for mice at least, that it can be delayed in very significant ways, that we can keep animals alive and healthy, cognitively active, and physically active for quite a long time.
The longevity field still has to fight a lot of myths, like the idea that extending lifespan is bad for the economy or even for civilization as a whole. You started a career in geroscience back in the 1970s. How have people’s attitudes changed over that period?
There’s a lot less progress than you might think. When I give a talk in front of many audiences, but particularly in front of audiences of non-scientists, the first or second or third question is, “Wait, what if everybody lived a long time, the economy would collapse, et cetera.” My response to that is, “If people stay healthy and productive longer, that, in general, is a good thing.”
If you think it’s a bad thing, then you might want to stop cancer research, to hand out cigarettes to children, to take insulin out of the pharmacies and seatbelts out of the car, because all of those are designed to keep people active and healthy and not dead for as long as possible.
It’s just as moral to try to develop pills that slow aging as it is to try to develop treatments for strokes, heart attacks, cancer, and diabetes. Both of those are a good thing to do. If we had been having this conversation in the 1800s, someone might have said, wait, you must stop! Because of all the science you’re about to do where you discover anesthesia for surgery, insulin for diabetes, penicillin to treat infections, the world is going to fill up with 50-year-olds, maybe even 60-year-olds!
What about the scientific progress? From many scientists I talk to, I get this mixture of optimism and pessimism: yes, we’ve learned so much, but we still know so little. What would you say?
As recently as the early 1990s, you could publish an article, there were actually several articles published in Nature proving that aging was so complicated that you could never slow it down by a single gene or a single drug or a single manipulation.
That was the prevailing scientific opinion among people who really thought about it and who were very smart. It’s now clear they were wrong. There’s now solid evidence in multiple species, most importantly mice, that some drugs, some diets, and some single-gene mutations can slow aging.
Once those discoveries are made, in addition to the famous work on calorie restriction, this attracts more people into the field because now that it’s clear something can be done to slow all of the aging process, that’s a reason to want to study it. The other thing that made a big difference is that many of these discoveries were made in animals that age very quickly, like the worm C. elegans and the fruit fly Drosophila melanogaster.

That makes it possible for graduate students, post-docs, and junior faculty members to get data in a year, rather than having to wait three or four years. That’s very attractive and it brings a lot of people into the field. It has led to many important discoveries in invertebrate aging of which a small, but not zero proportion also apply to mammals.
So, it’s nice to have an increasingly respected and productive field, where people are getting published in top-notch journals, famous journals. We now need to move further towards the point where research in mammalian and particularly mouse aging has a similar degree of attractiveness to the smartest people and the best money.
We’re not there yet. We’re a step or two closer to that, but it’s still an area where both the number of smart people working in it and the amount of money to support it are too small.
Yes, we’ve had a lot of success in mice, but many drugs that work in mice do not work in humans.
And many drugs that work in mice do work in humans. It would be silly to maintain that the percentage that work is zero, and it would be equally silly to maintain the percentage that fail is zero.
Most of the drugs that were developed for therapeutic effect in people were initially discovered by working on mice and rats. It would be nuts to say that every drug that extends lifespan in mice will do the same thing in humans, but the work in mice is a very important foundation.
Many of the pathways that are discovered, and maybe even some of the druggable targets that are first discovered, in the mice will serve in humans – maybe the same drug, maybe drugs of the same family, maybe drugs that target the same molecule, but through a different chemical grouping. It’s necessary to be neither insanely optimistic nor insanely pessimistic.
We do have a history of failures though, such as with Alzheimer’s, maybe because mice don’t really develop Alzheimer’s.
Yes, that’s true, but it’s important to recognize the brains of people and the brains of mice have a lot of things that are not in common. In terms of aging, if I tell you that I have an individual right here in front of me, in my office, that has cataracts, bad hearing, weakened bones, a poor immune system, and a relatively low cardiovascular system, you would immediately recognize that individual as old, be it a mouse, a dog, a horse, or a person. But you wouldn’t know if that’s a seventy-year-old human, or a 25-year-old horse, or a three-year-old mouse.
So, the effects that aging has on mice and on humans are – not in every case, of course, but in most cases – recognizably quite similar. And that’s true for cells that divide, for cells that don’t divide, for structures like the bones and the tendons that are mostly extracellular material. It’s true for complicated circuits, like neuroendocrine feedback circuits, it’s true for cognition.
There are just so many aspects – not all, but so many aspects of aging in humans, mice, dogs, chimps, et cetera that are the same. So, it’s very reasonable to expect that the drug that could block aging effects in all of those tissues in mice might also do very similar things in people.
But different species die in old age for different reasons. For instance, around 80% of lab mice die of cancer, I think.
The specific thing that kills the animal is of secondary importance when you’re studying the biology of aging. For instance, elephants die because their teeth wear down and they can no longer eat. When they’re 60 or 70, they have lost their last set of molars and they can’t chew food anymore. Mice, at least those that are used in aging research, indeed die mostly of tumors. People that eat a lot of fatty foods and watch TV, die mostly of atherosclerosis. In people that were alive a hundred thousand years ago, the most prevalent cause of death was probably breaking a bone and not being able to keep up with the group.
The point is not what is the specific cause of death in a specific environmental setting and in a specific species. The real question is what is it that postpones age-associated decline in bones, brains, the immune system, the sensory systems, the gut, and everything else for many decades in people, for a few years in mice, and for 20 years in horses. The factors that regulate the timing of the aging process, I would guess, is very similar in nearly all kinds of mammals.
Let’s talk about the ITP. Recently, there have been some very exciting results. Could you give us an update?
Of the two most recent interesting papers, one has to do with a drug called canagliflozin. This was published a couple of years ago. Canagliflozin is very frequently given to people because it’s good for diabetics. It doesn’t prevent glucose absorption, but it prevents very rapid increases in blood glucose levels, so you can see why it would be good for a diabetic.
It turns out that if you give it to mice, in male mice, it leads to about a 15% increase in lifespan and puts off at least five different kinds of non-cancer, non-lethal, non-neoplastic diseases in mice as well. So, it’s authentically an anti-aging drug.
It’s the second of the anti-aging drugs in mice that work apparently by blocking the highest glucose levels during the day. So, for cancer biologists, this ought to be very interesting. You’d really like to know how it is that you can postpone cancer by moderating the highest daily spike in glucose. In terms of humans, it would be really interesting to know whether canagliflozin and other fairly safe drugs that prevent extreme spikes in glucose might also block other kinds of age-associated lesions, independent of the benefits for diabetes.
The other paper, which I think was just submitted this week is one in which we tried a combination of rapamycin plus acarbose. Rapamycin works very well in male and female mice, while acarbose works significantly in both sexes but has a much stronger effect in males.
What we found is that when you give rapamycin and acarbose together, in the males, you do better than either rapamycin by itself or acarbose by itself. And that combination of drugs together gives male survival a 29% boost. That’s the largest percentage increase we’ve seen in males or females.
When you give acarbose and rapamycin together to females, they don’t do any better or any worse than on rapamycin alone. This is not too surprising because acarbose gives only a small effect in females. We expected it wouldn’t have a big boost over rapamycin alone in the female animals, and that’s what we found. So, the combination is the best thing we’ve ever had for any sex, although it is male specific.
Do we have any idea why most geroprotective drugs seem to work better in one sex than in the other?
While we don’t know why several of these drugs either work only in males or work better in males, there are several clues. Let me tell you about three of them.
One has to do with improvements in function like balance and grip, strength, and endurance on a rotating rod. Michael Garrett has studied several of these in mice treated with either 17α-estradiol, which is male-specific for lifespan, or acarbose, which males respond to better.
He finds that most of the performance measures are improved by acarbose and 17α-estradiol only in male mice. But there are a few that are improved even in female mice. In this way, you can begin to sort out what aspects of improved health are seen in both sexes and what aspects are seen only in males and presumably contribute to their lifespan benefit.
Similarly, people in my lab like Gonzalo Garcia have evaluated biochemical indices. Gonzalo has looked at enzymatic cascades in particular. He’s looked at two sets of kinases, one of which is really important in inflammation and the other in terms of protein translation.
When you look at the cascade that is important in inflammation, all the drugs, rapamycin, acarbose, or 17α-estradiol, cause benefits in that particular set of cascades in both males and females. So that’s not likely to be a key element of the lifespan control pathway because 17α-estradiol does not improve lifespan in females, it’s males only, and yet it does improve this kinase cascade.
The other kinase cascade, the one that focuses on protein translation, does show sexual specificity for 17α-estradiol in Gonzalo’s assays. Only the males benefit from 17α-estradiol. So, this is a hint that of the many cellular changes that these drugs produced, some, like the ones that lead to protein translation, at least show the same pattern of behavior as lifespan and may be important to lifespan. Others, like the inflammatory kinase cascade, respond to all the drugs, and that means that they’re probably not involved in the lifespan effect, which is male-specific for this. The more drugs we have that can be thrown into this analysis, the more likely it is that we’ll zero in on the specific pathways by which these drugs produce health benefits and lifespan improvements.
Our current search is to look for biochemical and physiological changes that are produced by all those drugs, as well as by four single-gene mutations that extend lifespan, and by caloric restriction. So, we have eight such changes so far – things that you can measure in young adult mice after drug treatment, or in the four mutants, or after calorie restriction. And they all change in the same way.
Most, though not quite all of them, are sex-specific for the 17α-estradiol treatment. We think that these physiological changes are our best glimpse as to what aspects of biology have to be changed, at least in mice, to get a lifespan increment.
This is also a link to human biology because you can ask, okay, I’m giving this drug to humans. Do any of these changes happen? If the answer is yes, that’s a sign that the drug may be very important in terms of controlling the human aging processes or at least links to the human biology and human diseases.
I think the ITP is a fantastic initiative. Why hasn’t it been massively expanded? Wouldn’t you want more money, more resources so that you can test more compounds, do more follow-ups? How would you improve the ITP if money wasn’t a problem?
Certainly, I agree with the idea that it deserves a lot more money. The Aging Institute has been pretty generous to us actually. For the first five years, each of the laboratories got half a million dollars a year plus indirect costs, and that was enough to allow us to do about five drugs a year.
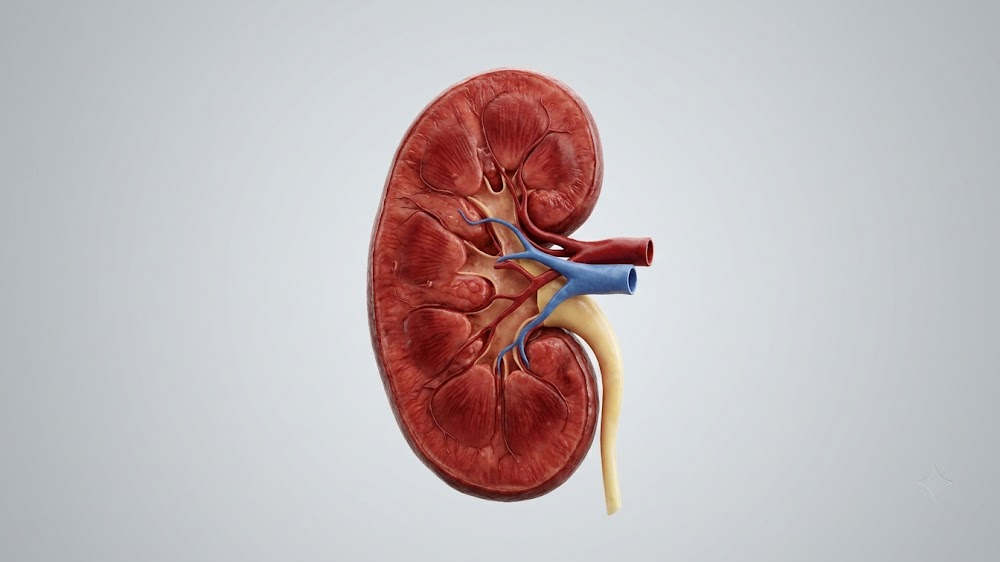
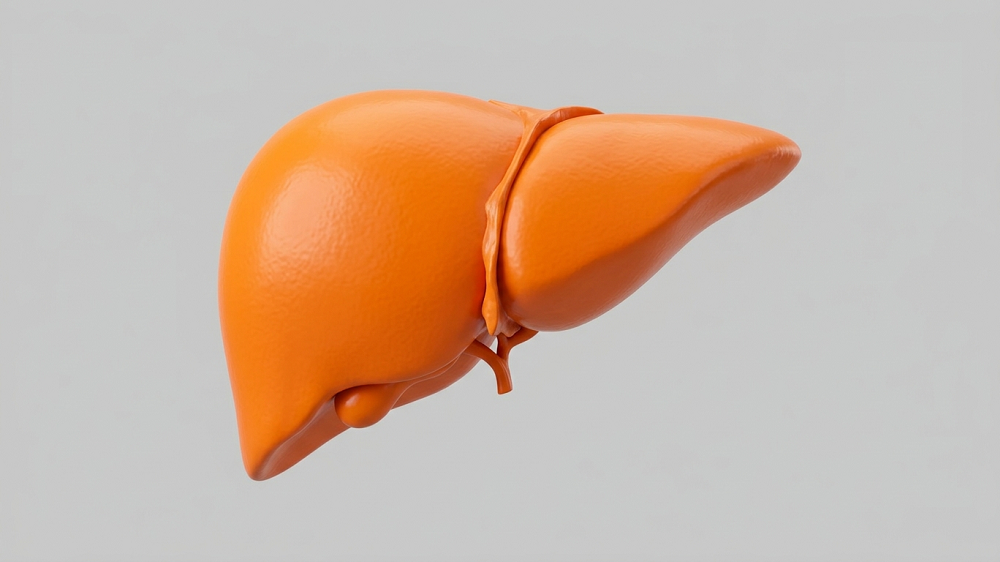
They were impressed by the work that we accomplished, and then after I think the 10th year, they doubled our budget. This allows us to do seven drugs a year and to do pathology on any drug that is a winner. It allows us to test in middle age some of the health outcomes that I mentioned to you like grip strength and balance on the rotarod. It also allows us to have freezers full of tissues from these mice so that if any lab in the world wants to know what that drug does to the liver or to the heart or to the kidney, we can say, sure. Here’s some tissues, see what you can find and let us know.
So, the program is well-funded. On the other hand, it’s easy to imagine things that could be done to accelerate discoveries, if more money were available. Some of the obvious things are, for instance, that we would like to know, if a drug starts in young adult mice and has a good effect, what would happen if you started it in middle-aged mice, would you get the same effect? We can do that now, but we have to wait five years before we get the first set of data, and that tells us whether we want to repeat it.
We also want to do dose-response curves. Each time we’d try a drug, we have to pick one dose and try it out. We hope we guessed, and sometimes we did guess, but it’s possible that the best dose of some drug might be three times higher or three times lower than the one we used. Having additional funds would allow us to do dose-response curves. The other thing that would be a lot of fun to do would be to evaluate mice that are known to be prone to specific kinds of diseases.
There are mice that get pathological changes in old age that mimic Huntington’s disease or Parkinson’s disease. There are some kinds of mice that in old age have cardiac problems, for instance, or specific problems in hearing. It would be lovely to know whether these drugs that extend lifespan in our heterogeneous mice would also slow down this or that disease. Those are obviously critical steps towards making this important translational jump to human diseases.
That’s already being done. My lab with Roger Albin looked at a Huntington’s disease model, and several labs in Texas have looked at Alzheimer’s disease models, et cetera, but the money that comes to the ITP labs mostly has to go towards screening new drugs, which is our specialty. So, we don’t have tons of money left over to apply the same kind of analysis to mice prone to individual diseases of importance.
Do you think the ITP with its “instant reproducibility” should become a blueprint for other studies, maybe outside the context of aging?
I’m sympathetic to that idea, but of course you understand there’s a balance between costs and outcomes. Many experiments are done in a single laboratory, and if they’re published, and people find them exciting, then other labs often do try to replicate them. One of the reasons that with the ITP, we thought it would be better to do the replications from the start, is that these are aging studies, and from the time that mice are born to the time the last one dies, it takes three to four years. If you waited four years to get your result and only then began your replication, you wouldn’t have a solid confidence for eight years. So having replication built in from the start speeds the process up quite dramatically.
The other reason is that each of the three sites had some uncontrolled variables that make it different from the other sites. We do our best to control things – we buy the food from the same kitchen, bedding from the same place, we use the same kind of mice bred in the same month of the year. And despite all the precautions there are site-to-site differences.
The mice at Michigan are always lighter for both sexes than the mice at the Jackson Labs or Texas. The females from all three sites always live the same period of time, but the males from Michigan almost always live longer than the males from the other two sites. The reasons for this are unknown. They may have to do with the temperature in the room or things in the water that we can’t smell or taste, but the mice can.
But if all three sites produce a good, strong effect, we can be fairly confident that the result is reliable and would be seen at any well-managed animal colony using the same drug and the same kind of mice. Often though, we’ll find a drug that works at two sites, but not at the third site. And then we would publish that, and we think it’s important and interesting, but we don’t have the same degree of confidence that we can always replicate it. Those are the advantages of replication.
Human lifespan makes studying anti-aging interventions in humans even harder. How can we even measure aging in humans? What are your thoughts on the design of human trials like TAME?
That’s an interesting set of conundrums, and it’s partly scientific, partly financial, and partly legal. The smart way, if you are in charge of the universe of designing an aging trial, is taking an awful lot of people when they’re 30 or 40, have them take the drug you are interested in, and then come back in 60 years.
No one’s going to do that, it’s very expensive. And drug companies really don’t want anti-aging drugs, because they don’t want to invest hundreds of millions of dollars and not get an answer for 20 or 30 years. Also, the FDA explicitly forbids aging as an endpoint for any of their drug trials.
A group of 11 research universities put together a study that attempted to meet some of those obstacles. For instance, they defined an endpoint that is not getting diabetes, not getting Alzheimer’s, not getting cancer, not having a heart attack, not having a stroke – I don’t know the details of it.
And the FDA will take that. As long as you don’t call it aging, but prevention of many diseases, the FDA will say, sure, go ahead. But it’s still a 50-million-dollar trial that will take a minimum of five and more likely eight to ten years.
And even then, you have to start with people who are old, in their sixties, so that the number of people who reached the defined endpoint within a five-to-ten-year period will be high enough to give you statistical power. You are hoping that the drug you’re testing, which is in this case, metformin, will work really fast, even on people who are already pretty old and maybe sick.
That could be the case. But it need not be, it could be that some drugs will work in people, but only if you start giving them to people when they are 40 or 20 years old. The people who put the TAME trial together are hoping that’s not true, at least for metformin.
But if this is true, and we need much more time, they might reach a conclusion that metformin doesn’t work when, in fact, it does.
Yes, if metformin works at all, it may be that it would work only if you start in 30 and 40-year-old people. We now have data on four drugs that gives us a clue of the possible scope of answers to this kind of question. Astonishingly, rapamycin works just as well in terms of increases in median lifespan, whether you started at 9 months or at 20 months of age [in mice]. I thought that was very unlikely to be the case, but I was wrong. It is fully active, even if you start as late as 20 months of age.
For acarbose, the answer is if you started in midlife, you get half the effect. For 17α-estradiol, if you started at 16 months of age, which is early middle age for mice, it works just as well as if you started it at 9 months of age. For canagliflozin, we don’t know yet. We started the study, but we don’t have enough deaths to say for sure. These mouse studies, of course, don’t perfectly predict what you would see in people, but they give you an indication of the range of possible answers. So, of the three drugs we have so far, two work quite well if started in middle age.
They’re hoping metformin too works just great, regardless of how late you start. Metformin does not extend lifespan in mice, but nonetheless it may do so in humans.
That was one of the most high-profile failures in the ITP history, I think. Do you have any guesses about why that happened? Would you like to go back to metformin and maybe test it differently?
Each year, we get 10 or 15 good ideas to test. And if someone said, hey, would you mind going back and testing metformin again, they’d have to make a pretty strong case that it might work the next time. Maybe they have a different dose or a different formulation, or maybe they want us to give it three days out of every seven or something.
They could make a case that retesting metformin is a good idea. My hunch is that it’s not worth retesting. The reason is that it was actually tested twice – once by the ITP, and then once before us by Rafael de Cabo. He found that it did not have any effect in female mice. In male mice, he said it had a small but significant 5% effect, but I don’t put much faith in this conclusion, because he did not use the standard statistical test.
On the other hand, we do have some robust data that metformin benefits humans.
The epidemiological data are quite provocative, I agree. It’s very encouraging. On the other hand, it’s an observational study, and it’s a post-hoc analysis, and those are known to be misleading sometimes. The epidemiological data in humans is pretty strong and make a reasonably good case that you should pay more attention to metformin in humans.
Some of the drugs that the ITP is testing, and I’m thinking in particular canagliflozin, are already being used by tens or hundreds of thousands of people. They are now a mainstay of diabetes therapy. Probably the quickest thing you could do is come back to people who have diabetes and who’ve been on these drugs for a decade and ask, “How about cataracts? How about hearing? How about bone breaking? How about a lot of stuff that’s not clearly related to diabetes, but is clearly related to aging?” If canagliflozin and other SGLT-2 inhibitors are slowing the aging process in humans, then you would predict that the drug not only protects you from diabetes but could protect you from a lot of other problems that occur in old age.
If this is the case, we have a real anti-aging drug. I think that would be at least as informative as the TAME trial. Similarly, acarbose is a mainstay of anti-diabetes therapy – not in America, but in many Asian countries. And one could ask the same kinds of questions in Asian countries for people who have been on acarbose for a good part of their adult life.
That would still be a population study.
I agree. I like controlled trials. But if you’re asking for a 30-year controlled trial, you’re going to get some pushback.
What are the major takeaways from the ITP as of now?
We’ve already talked about some of it today. One major point is establishing definitively that you can slow the aging process down by putting something in the diet. It refutes the commonly held notion that you cannot change the aging process.
The data is now overwhelming that aging can be slowed ad you do that by extending healthy lifespan. The evidence does not support the nightmare scenario where you get people or mice that just won’t die – they’re demented, they’re in pain, they’re in terrible shape, but they just won’t die. The drugs the ITP has tested do not do that at all. Instead, it’s keeping them healthy longer, and that’s why they stay alive longer.
The second point is that it gives you tools for working out the mechanisms by which aging can be slowed. We now have at least four genes, four drugs, and two diets that consistently, reproducibly increase mean and maximum lifespan in mice. So now, you can, for the first time, begin to ask questions through an entirely new paradigm of how to learn about aging. Up until maybe the 1970s, 80s, or 90s, the way you did aging research was that you made a list of things that differentiated old from young mice, rats, or people, and then you fantasize that, oh, this one looks really important, maybe that’s the cause of aging. The new approach is much smarter: you compare young animals that are either normal or that have been treated in a way that makes aging go more slowly.
If you want to know what the cause of slower aging is, you don’t have to wait for aging to take place. You can figure out what is going on at the very earliest parts of the lifespan that slows the aging process down. That’s a whole new paradigm and a very powerful research design.
The third point that’s probably worth mentioning: each of these drugs has a target. Sometimes, we don’t know what the target is. For rapamycin, the target famously is an enzyme called TOR, the target of rapamycin, but we don’t know yet whether the anti-aging effects of rapamycin are accomplished by a change in the brain, in the pineal, in the brown fat, in the white fat, in the liver, or in the arteries.
We have to define the specific cell types that rapamycin influences in ways that create broad healthspan and lifespan benefit. That’s a critically important next step that we can’t take without the drugs. The same is true for acarbose and canagliflozin. They strongly indicate that at least in males, you can slow many aspects of aging, including death by preventing daily surges in glucose. That’s an opening. Why is that? Why is it that the daily surges and glucose are a key element in males for timing the aging process?
You can also begin to ask similar questions about sexual specificity. We have several drugs that seem to work better in males, and we’d like to know what it about the physiology and pathobiology in male animals that makes them susceptible to something that these drugs address.
The last point is that the ITP serves as a foundation for studying possible preventive medicines that might work outside the laboratory environment in people.
The critical next step probably is dog research. Matt Kaeberlein, Dan Promislow and their colleagues are treating dogs with rapamycin, and they’re starting with middle-aged dogs. It won’t take very long – probably five to seven years to get some lifespan data. If they prove that rapamycin at the dose they’ve chosen extends healthy lifespan of dogs, that will be a major breakthrough, because if it works in mice and in dogs, you better think it’s going to work in people too.
If I were in charge and had lots of grant money to give out, I’d want to be sure that there were more dog studies that evaluated other anti-aging drugs that the ITP had developed, to see whether they also had a beneficial effects in a species that’s much larger than mice and has the same kind of genetic heterogeneity that humans have.
By the way, why dogs and not, let’s say, small monkeys?
Oh, that’s easy. Dogs live with people and there are millions of them. So, you don’t have to build a whole building and fill it with experimental dogs. What you do is you tell people, would you like your dog to have a 50/50 shot at getting a medicine that might extend its lifespan? The dog stays with you, lives with you, eats dog food, except we’ll put our drug into it. And you your dog also gets a free checkup once a year. So, it’s really easy to sign up volunteers for the study.
What do you think about other types of anti-aging interventions, such as gene editing and cellular reprogramming?
For gene editing, you’d have to ask a couple of questions. What genes are you going to edit, what cells are you going to edit your genes in, and what makes you think it would do any good? Currently, we don’t know of any genetic change you can do in a specific cell type that slows aging in any mammal. There are lots of exciting, hot, new techniques that in principle could do cool stuff. The notion that they are the secret to slow aging is, as of now, a good point for science fiction. I’m eager to hear from someone who claims they know what cell type they need to edit and what gene they need to edit in that cell type.
Same for cellular reprogramming: why do you think this is going to be good for you? Are you planning to reprogram the cells in my brain, or in testes, or my bone marrow cells? And what are you going to reprogram them to do? Are you pretty darn sure they’re not going to get cancerous?
It’s a valid point, of course, but theoretically, how far could these techniques get us?
These are broadly hyped and potentially very powerful techniques. What is needed is a clear idea about how you can apply these dynamite techniques to ask and answer a well-defined hypothesis about aging. One can imagine such a hypothesis – for instance, that reprogramming a specific cell type in the hypothalamus so that it doesn’t show inflammatory change would slow the aging process down.
Then, one could, in principle, define a viral vector that specifically infects that kind of hypothalamic cell and modifies NF-κB-dependent processes to block inflammation in the hypothalamus. If that’s technically feasible, it could well be very informative.
Plans of that sort, where you have a specific target, a specific genetic manipulation, and at least a plausible model for why that leads to a health benefit, would be thrilling. But simply waving your hands and saying, oh, I’m going to do gene editing, doesn’t substitute for a good plan to test a hypothesis.
So, maybe the ITP could provide the insights that those areas need?
Yes, information about what genetic targets in what cell types need to be manipulated to slow aging. That would be really nice to know, and sometimes, the work in the ITP and other labs that are stimulated by ITP results can move us several steps in that direction.
To give just one example, Gonzalo Garcia has come up with a really nice observation that both in drug-treated slow-aging mice and genetically modified slow-aging mice, a lot of messenger RNA is translated by a special process: – cap-independent translation. That suggests a hypothesis: if you have a drug that can turn on cap-independent translation, that drug might be really good for you. Or if you’re into gene editing and you know what gene controls cap-independent translation, you can imagine a way of modifying cells to promote cap-independent translation.
The ITP could lead us to a greater understanding of the aging process: what controls its rate, what cellular and genetic targets are plausible for genetic engineering and cell therapy? That’s one way in which the work we’re doing could contribute to the overall endeavor.