Professor of Genetics at Harvard Medical School, a veteran geroscientist, and a serial entrepreneur, George Church hardly needs an introduction. While we are always happy to discuss the present and future of geroscience with him, this interview focuses on the two gene therapy papers that he recently co-authored, which drew a lot of attention due to their spectacular and surprising results. In this interview, Prof. Church interprets these results and gives his opinions on a range of longevity-related topics, such as cellular reprogramming and supplements.
Two recent papers that you were involved in have caused quite a buzz in the longevity community. Let’s start with the telomerase and follistatin paper. My first question is, why didn’t you try their combination?
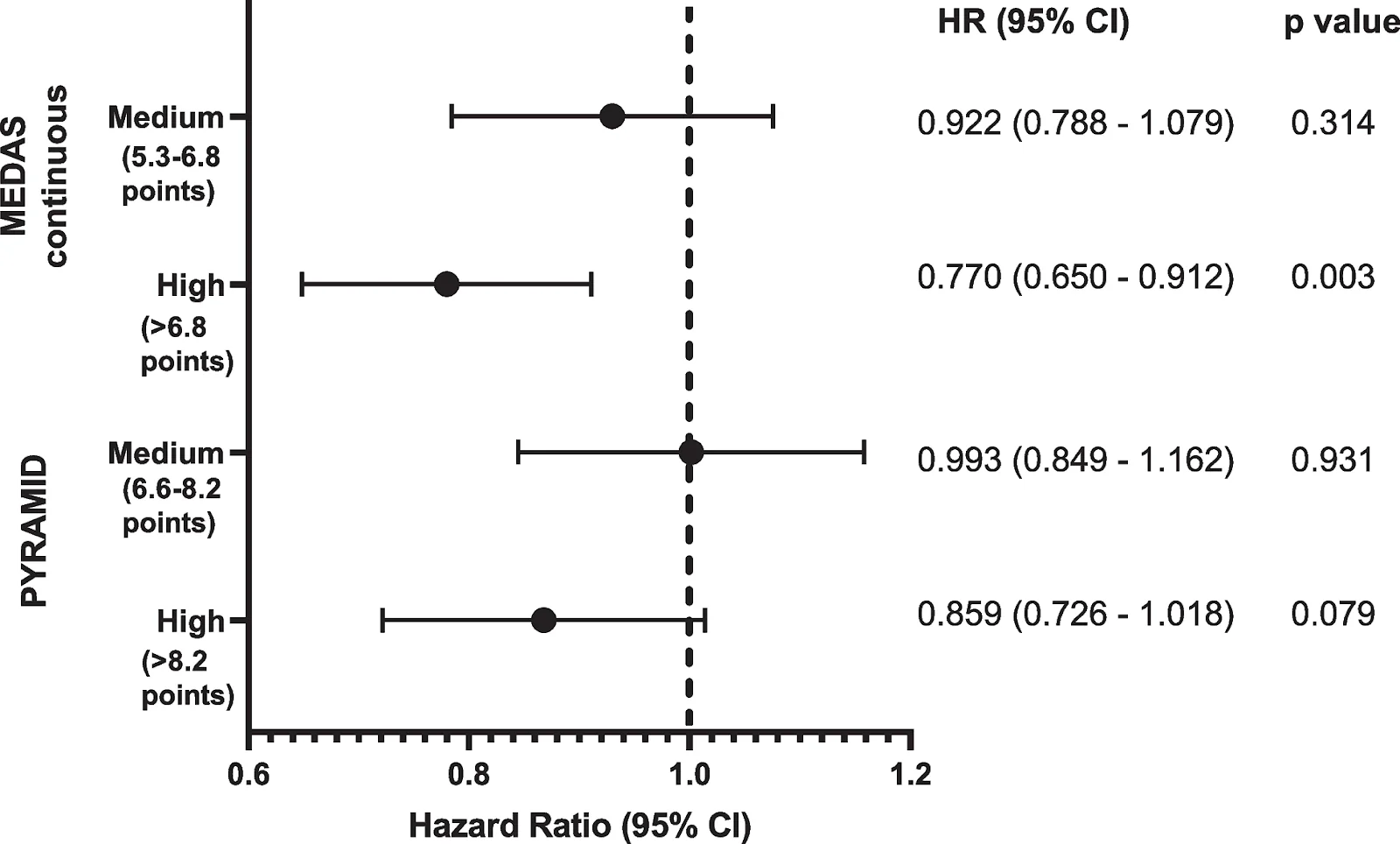
That’s a good question, especially given that we were using this huge vector. The whole point of cytomegalovirus is that it can package a lot more. I think it’s just that these experiments are not cheap, including the mice. What is distinctive about both papers is that they have a Kaplan-Meier plot, and those are more expensive than just showing some reversal of physiological decay or of an age-related disease. Both groups are thinking about combinations, they just haven’t done all of them yet.
Do you have a prediction about the possible effect of a telomerase and follistatin combination?
Those are fairly independent pathways. There are arguably ten major pathways of aging, and I think you need to get all ten pathways if you want to impact longevity beyond a certain point. It’s been estimated that doing just one of those ten pathways might get you two years of life extension in humans and maybe a few months in mice. But, if you develop something that impacts multiple pathways, some combination drug, that will be very interesting to see when we get to that.
Follistatin, in particular, is a rather unusual choice of an anti-aging treatment, but it did deliver fantastic results. Do we have a mechanistic explanation for that?
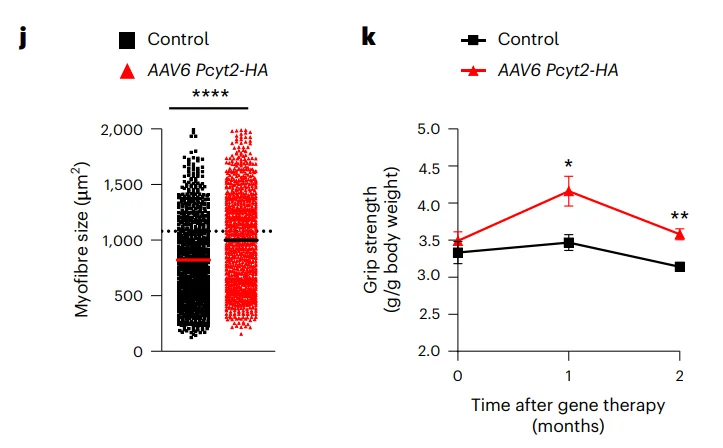
I think the original explanation is that cachexia is muscle wasting that accompanies a lot of aging. You could argue that’s just a late-stage thing, and to do real aging reversal you’d want to get to the earlier stages, or you could argue that this is one of the ten pathways, and you want to have that part of a combination, but the answer is that it’s still not clear why it works in itself.
Cachexia accompanies cancer, the main cause of death in lab mice. Could this hint at an answer?
It’s one of the mechanisms that cancer can cause death by, but there are others. Cancer can cause death by making it hard to breathe. It can screw up almost any physiological process, depending on where it starts and where it metastasizes, but muscle wasting can be a product of fairly normal aging without cancer as well. Seems like it’s a nice tool to have in your toolbelt, but I admit it is a little surprising that it works by itself.
Telomerase is also an important factor in cancer. Do you think telomerase treatments can have adverse effects in humans that we don’t know about yet?
First, there are effects that we do know about. If you give a long steady dose, it will increase the probability of tumorigenesis. It also sets you up for additional genetic and epigenetic changes that can make an even more serious cancer. But, if you give intermittent doses, it’s like with Yamanaka factors in the second study: if you give too much, cells could go back too far in time. So, I think it’s all about moderation.
The other thing that was done in germline experiments, or earlier animal models, is to use specific safeguards against cancer so that when you then introduce the telomerase, you can express it at higher levels and for longer periods of time. For instance, messing around with the p16 and p53 pathways, i.e., having extra copies of tumor suppressors, can protect you against certain forms of cancer and allow you to use things that would otherwise put you at risk.
The results of this paper were spectacular, and it got us all very excited, but the sample size was small. Are there any plans for a bigger follow-up study?
It’s all about resources. Both those companies are scrappy, young, underfunded, but they make up for it in creativity and delivery. Those are the only two gene therapy papers that I know of that show significant Kaplan-Meier plots. And I really like the gene therapy approach that they share, because you can target alleles, splice isoforms, and gene family members (paralogs). This is something that’s very hard to do with small molecules, but very easy to do with either protein or gene therapies.
The slight advantage of gene therapies over protein therapies is that you have a few ways of rigging the specificity. You can have specificity of the vector, specificity of the nucleic acid, and finally, specificity of the protein. With proteins, you only have the latter.
The only thing I don’t like about gene therapies is the cost, but one positive side of the COVID dilemma was that we tested five different vaccines that were formulated in the form of a gene therapy on billions of people and got the price down to as low as 2 dollars for one of those five. It’s a whole new ballgame now if you have a large market, and I think pandemics and aging are the largest.
Let’s move to the OSK paper that came out of Rejuvenate Bio. What is the importance of it? Was it the simultaneous delivery of three factors in one vector or something else?
We had already done three genes at once at my lab and Rejuvenate Bio. Those were different from the OSK Yamanaka factors. The latter are transcription factors that need to be delivered to the nucleus, while the other set that we had, TGF beta receptor 2 in soluble form, Klotho, and FGF21, could be soluble, secreted, hence they could go into the interstitial, intercellular matrix, or they could go out into the blood. In a certain sense, it requires less delivery, or less ubiquitous delivery of the nucleic acid, because then the proteins do some of the delivery.
That’s the fundamental difference between those two. The significance of using the OSK is that this was the best example that we had of rejuvenation, as opposed to just fighting the symptoms of some age-related disease, making your muscles hurt less or something. This was truly rejuvenating ancient cells all the way back to embryo.
So, if you can control that, dial it up and down, it has some promise. This encouraged several groups, including Altos Labs, which I worked with briefly (many of my alumni are core members of Altos Labs). It is also one of the things that encouraged our collaboration with David Sinclair.
But, except for Rejuvenate Bio, so far, all of those are fairly distantly related to longevity. They might cure something like a crushed optic nerve, but damage to the eye is not aging, certainly not longevity. Now, this wasn’t merely the Kaplan-Meier plot where you could see extended life. The injection occurred at the age of 124 weeks. For a mouse, it’s very late in life. Half of the mice in the cohort are already dead at that point.
That was indeed a very interesting design. I think those were the oldest mice in any study that I’m aware of. What was the rationale behind that? What were you trying to prove?
The logic behind this was that if you do it as late as possible and show that it still has an effect, that’s very encouraging. It broadens the market, that’s one way of thinking about it, but it also tells you that you are really getting a reversal as opposed to delay, and that’s a fundamental difference. Many drugs that are considered cures are reversing something, but people somehow put reversing aging in a special category, like breaking the sound barrier. However, just like breaking the sound barrier was easy enough to do once we figured it out, reversal of fundamental epigenetic programs that are a natural part of development and aging starts looking easier than it originally did.
Do we understand how cellular reprogramming improves health and longevity?
There have been two major camps in aging since long ago. One says that aging happens due to damage, to proteins, lipids, RNA, and DNA, and that you have to go in there with your repair kit and fix it as a therapist. The other camp says that it’s all epigenetic, and that if you convince the cell that it’s young, it will get its own toolkit out and start repairing as much as it can. Some things are beyond repair. If you delete all copies of a tumor suppressor, that’s not something a young cell can repair. But most things are fixable with epigenetics – at least, that’s how the second hypothesis goes.
I believe in a hybrid model. I think most of the work can be done epigenetically. A surprising amount of it can be done via the bloodstream, but probably not all of it. Then, there’s a residual amount that you can fix with the Yamanaka factors and another residual amount that you can fix by restoring genes.
Since we do the epigenetic reprogramming by adding in genes, it’s not that fundamental a difference between adding in genes that will go into the blood, adding genes that will reprogram the nucleus, and adding genes that are missing, like tumor suppressors. In a certain sense, they are all addressable by multiplex gene therapy. That’s why being able to either use multiple rounds of dosing or to have bigger vectors will become increasingly important.
Given the rising popularity of partial reprogramming, what is its overall place in the longevity landscape?
I think there are subtle but important differences between anti-aging drugs and drugs that improve biomarkers in the way that statins improve cholesterol. That doesn’t mean such drugs increase longevity, just that they improve this one biochemical. It could actually hurt you; for instance, it could improve cardiovascular chances for some subset of the population, but for another subset, it could hasten muscle pain.
So, affecting biomarkers is one thing. Reversing diseases of aging is different. You could do it just by addressing that particular disease, or you could do it more broadly, affecting multiple diseases. You might get FDA approval for one of them, but it’s actually affecting multiple ones, and maybe acting preventatively. Say, there might be a cure for muscle wasting that helps prevent a variety of diseases.
Finally, you’re really at the core of aging when you reprogram shared elements – with good feedback systems that already exist in the body or with feedback systems that you introduce as part of the therapy.
I understand that the combination therapy with the three soluble factors you mentioned earlier is already in clinical trials in dogs. Do you have any preliminary results?
The thing about clinical trials is that you ideally minimize talking about them before they’re done. This is a second animal trial. We did it in mice already, and it looked good. With the dogs, it’s not just an animal trial, it’s also a product, a veterinary product. I understand that some of the pet owners involved have said good things, but, of course, you have to be very cautious with anecdotal evidence. There could be wishful thinking, placebo effect involved. These are randomized clinical trials, so you really won’t know until you break blinding at the end.
But the nice thing about veterinary clinical trials is that they’re typically much faster than human clinical trials. COVID-19 trials were an exception, 12 months, but most human clinical trials last about 10 years, and most veterinary trials are more like 18 months. So, we’ll know soon.
Still, the endgame is delivering those therapies to humans, and you will need a human trial anyway.
We could have gone straight from mice to humans, but we decided to create a veterinary product. People spend a fair amount of money on end of life in dogs, they even clone dogs, but of course, a clone is not what you want. So, it’s valuable to have two or more animal preclinical trials before you go into clinical trials. Even though we’re looking into specific diseases, for instance, the mitral valve disease in spaniels, it’s not limited to that. We hope that it will hit many different diseases. We’ve tested it in about five or six different diseases already, in mice and dogs.
To move this into humans, you use the same genes. You might use a human version of them, just like you might switch from mouse to dog genes, but other than that, mode of delivery. You might also change the vector. We use AAV vectors, which is the oldest approved vector and one of the most popular ones. However, dogs have natural immunity to most of the popular AAV vectors.
That’s clearly solvable. It’s even solvable for redosing. One of the advantages of certain gene therapies is that you don’t have to redose. You might have a once-and-done, lifetime expression, but a couple of my companies are working on improving the AAV delivery. Shape Therapeutics and Dyno Therapeutics are both making quite a progress on this. So, you can make radically different capsids that have radically different tissue tropisms and immune evasion. You basically make viruses that don’t exist in the wild or in previous pharmaceuticals, and you can probably keep generating those things for quite a while.
First, with dogs, you can get a lot of feedback on how to maximize your chances of success. Second, it’s a product, so you can make a profit on the first product to pay for the development of the second product. Human clinical trials are expensive. You could do it by investment, but that would result in dilution of the original founder’s stake in it.
Also, dogs are just a particularly good intermediate between mice and humans, because they live in a human environment, they have some compatible behavioral traits and eating habits. You can identify personality changes, subtle behavioral changes that you don’t identify in mice, because you don’t cohabit with them like you do with dogs.
None of this means that we couldn’t go directly to humans, it’s just that failures in human trials can put a damper on the whole field. I hope that all people in the field do their homework and don’t rush trials, because they can screw it up for us. We’re trying not to screw it up for them, and, hopefully, that will be reciprocated.
By the way, you do have the reputation of being a serial entrepreneur. How are your numerous startups doing? Are you still bullish about longevity biotech?
I co-founded 46 companies. I’m on the scientific advisory board of a few more. Seven of them are related to aging reversal and longevity. They all involve pre-clinical and clinical trials. Some are on nutritional supplements, which you can sell without FDA approval, but nevertheless, they’re doing trials, and that’s a strong preference for my involvement in a company: they have to be willing and planning to do, and also have the resources to do, clinical trials, even if not legally required.
I think the whole field is very healthy economically and scientifically. We have passed through multiple “valleys of death”. We’re now in the solid science phase, and this field is going to be very impactful, maybe more impactful than any other pharmaceuticals in history, including even antibiotics, because our very ability to fight off diseases is age-related. Almost every single form of human morbidity and mortality has an age-related component to it. If you want to have a pleiotropic effect on many different diseases, this is the way to go.
So, it seems like a good choice. It also has a good chance of being cost-effective enough. I’m not promising this on behalf of any of my companies, but since manufacturers of COVID vaccines can get gene therapy to be as cheap as two dollars a dose, others can do that as well, and that’s important to me from the standpoint of equitable distribution of technology. It’s not about “can we develop a billion-dollar drug?” Rather, it’s “can we distribute it to everyone who needs it?” And the same way that we got the price of DNA sequencing down seven orders of magnitude, 20-million-fold, I think we need to at least consider that for every new medical technology.
Speaking of supplement companies, you are involved with two of them, NOVOS and Elysium.
Yes, and I was referring to them when I said they’re willing to do clinical trials even though they are not required to do so.
This is commendable, but they are already selling supplements. Do you think it’s not too early to be marketing anti-aging stuff to people, considering the state of the research?
I think we need to be very cautious about supplements that do not go through clinical trials and also about what trials they went through. If you do a clinical trial on ALS or some other degenerative disease, then that’s all you’re addressing. You’re not addressing the broader population. And, you have to be cautious, because some supplements have been shown to be detrimental if taken for prolonged periods in particular individuals that may have a pharmacological predisposition.
For example, some encourage cancer. And that’s true for all things you put in your body. Just because your grandparents got away with it doesn’t mean that it’s going to work, especially as we now have a more complex environment with a lot of other toxins.
Also, because we’re staying employed later and later in life, we need to reevaluate the things that were generally recognized as safe. So, I’m very enthusiastic about testing and retesting things that have been accepted without formal clinical trials. There’s a lot of placebo effects, long-term effects, and wishful thinking that we need to put to the test. I hope those two companies continue along that pathway.
So, you think they’re being prudent enough for you to associate your name with them?
I’m a scientific advisor. We shouldn’t ignore companies that need advice, otherwise, they will be operating without advice. As long as they’re doing clinical trials, they’ve got my attention.
Which directions in geroscience are you personally excited about at the moment?
I think we really need to hit all ten pathways and think about their interactions. In my opinion, the easiest way to go from a hypothesis or a pile of molecular data to a therapy is via gene therapy. In a couple of weeks, you can essentially go from a new research article to a therapy and get that therapy into mice.
So, I’m particularly excited about those directions. We don’t know yet whether it should be cell-autonomous or something that spreads through the blood, probably both. I’m excited about the possibility that this could be low-cost for equitable distribution. All those things are clustered around this multi-drug therapy, which I think is fairly well-validated in other cases: multi-drug antibiotics, multivalent antivirals, cancer, some other diseases as well.
So, you think we’ve accumulated enough knowledge to start moving in the direction of combination anti-aging treatments?
Yes, except you have to test them all in vitro and in animal models, in the exact formulation that you want to use, even if the ingredients were already tested individually. Sometimes with gene therapy, you can’t test the very exact molecular form, because there’s a difference between the animal form and the human form. But, other than that, everything should be as close as possible.
In one of your recent talks, you said something, and I hope I’m not misinterpreting, about cell therapies being superior to gene therapies.
It’s not either or. Cells can deliver genes and proteins. The advantage of cell therapy in principle is that you can debug the gene therapy ex vivo and then deliver it. For example, if you apply gene therapy in vivo to a billion cells, you’ve got a billion chances for genetic mischief, but with cell therapies, especially clonal cell therapies, you can debug it and make sure this is genetically and epigenetically what you want, that all cells are basically identical.
The downside of it is that many cells just don’t deliver very well. For example, neurons are highly connected. Maybe you can deliver a few neuronal stem cells, including into parts of the brain that normally don’t have neuronal stem cells, but you also want to keep most of your connections intact and not replace them with some generic neuronal progenitor. The bottom line is that there’s room for a lot of different strategies and innovations.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.