DNA is a critical part of cells, the instruction manual for building them. While DNA is well protected within the cell nucleus, damage does occur; therefore, DNA repair is absolutely essential for cell function, cell survival, and the prevention of cancer.
The good news is that cells can repair damaged DNA, but the bad news is that this ability declines with aging for reasons that are not yet fully understood.
An exciting new study by researchers led by Dr. David Sinclair at Harvard Medical School shows a part of the process that enables cells to repair damaged DNA, and it involves the signaling molecule NAD. This study offers insight into how the body repairs DNA, how DNA damage relates to aging, and why our repair system declines as we age. Before we get into the new research study, let’s take a look at how DNA damage relates to aging and what NAD is.
Genomic instability: a driver of aging?
The stability and integrity of our DNA is challenged on a daily basis by various external physical, chemical, and biological agents as well as by internal threats such as replication errors and reactive oxygen species.
Some aging theories, such as the Hallmarks of Aging, implicate DNA damage as one of the primary driving processes of aging, as it contributes to genomic instability [1]. Various premature aging diseases such as progeria are the consequence of accumulated DNA damage; however, the relationship between progeria and normal aging is as yet unresolved. This is partly due to the fact that the different progeroid syndromes only manifest certain aspects of aging seen in normally aging people.
What is NAD?
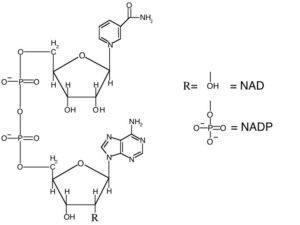 Nicotinamide adenine dinucleotide (NAD) is a dinucleotide, meaning that it consists of two nucleotides joined by their phosphate groups. One nucleotide contains an adenine base, and the other contains nicotinamide. NAD is found in an oxidized form and a reduced form, abbreviated as NAD+ and NADH respectively.
Nicotinamide adenine dinucleotide (NAD) is a dinucleotide, meaning that it consists of two nucleotides joined by their phosphate groups. One nucleotide contains an adenine base, and the other contains nicotinamide. NAD is found in an oxidized form and a reduced form, abbreviated as NAD+ and NADH respectively.
As part of its role in metabolism, nicotinamide adenine dinucleotide supports redox reactions, which involve moving electrons from one reaction to another. The transfer of electrons is the primary function of NAD, but it also has other roles.
Found in every cell in our body, NAD helps to suppress genes that accelerate the aging process and is a fundamental part of our metabolic system. NAD is associated with the sirtuins, which are closely linked to longevity in mammals and other organisms. Its control over cell damaging oxidation is also well documented. NAD declines during the aging process due to being actively destroyed by inflammatory signaling, as shown in a 2013 study by Schultz and Sinclair [2].
So, what’s the big news?
This new study demonstrates a previously unknown role for the NAD signaling molecule as a master regulator of protein-to-protein interaction during DNA repair. It also gives us valuable insight into why the body’s ability to repair DNA damage begins to fail as we age [3].
These experiments conducted in mice demonstrate that treatment with a NAD precursor known as nicotinamide mononucleotide (NMN) can mitigate and resist age-related DNA damage as well as the damage resulting from exposure to radiation. While there is no guarantee that these results will translate from mice to humans due to differences in biology, if they do, potential therapies may be the result.
Building on previous research
David Sinclair and his team already demonstrated that NMN can extend the lifespan of mice in a previous study [4] and reverses loss of mitochondrial function with age [5]. The team began this new study by examining the various proteins and molecules that they believed were involved in the aging process.
They knew that NAD, whose levels fall with age, increases the activity of the SIRT1 protein (one of the Sirtuin family) and can delay some aspects of aging, extending the lifespan of yeast, flies, and mice. They also knew that SIRT1 and PARP1, a protein that is involved in DNA repair, both consume NAD during their activation.
The team also looked at a protein called DBC1, a common protein found in humans and many other organisms from bacteria upwards. Studies had shown that DBC1 is able to inhibit the activity of SIRT1, so they believed that it might also influence PARP1, given their similar roles, and wanted to see if it was connected to NAD. It turns out that they were correct, and the study revealed this link.
The research group tested the relationship between the three proteins by measuring protein-to-proteins interaction within human kidney cells. They discovered that PARP1 and DBC1 actually bond strongly to each other, but when NAD levels increase, that bonding is reduced.
Simply put, the more NAD in a cell, the fewer bonds DCB1 and PARP1 can form, freeing up PARP1 so it can repair damaged DNA. They also took this further, inhibited NAD and noted the number of DBC1 to PARP1 bonds increased. This shows that reduced levels of NAD strongly influence the ability of cells to repair DNA damage.
These findings suggest that as NAD falls during the aging process, the less NAD there is to prevent DBC1 and PARP1 bonding, which is harmful to DNA repair. The result of this ultimately causes DNA damage to go unrepaired and accumulate over time, leading to cell damage, mutations, loss of tissue, cell function, and organ failure.
Getting down to the nitty-gritty
That in itself was interesting enough to have discovered this previously unknown function of NAD, but the researchers wanted to understand exactly how NAD was doing this. To find out how NAD prevents DBC1 from bonding with PARP1, they examined a region of DBC1 known as NHD.
NHD is a pocket-shaped structure common to around 80,000 different proteins in a huge number of species, and its function has been a mystery to scientists. The team showed that this NHD region is a NAD binding site, and in DBC1, NAD binds to this region and prevents DBC1 from bonding with PARP1 to prevent DNA repair.
Interestingly, NHD is so common in across species it suggests that this NAD binding might play a similar role preventing harmful protein interactions in many species, including humans.
Moving to mice
Next, the researchers treated old mice with NMN, but before they did they checked the protein levels in the mice. As predicted, the old mice had lower levels of NAD in their livers, as well as lower PARP1 levels and a larger number of bonded PARP1 and DBC1 proteins.
However, once given NMN in their drinking water for just one week, the old mice showed significant improvement in NAD and PARP1 levels. Tests showed the NAD levels in the livers of the old mice increased similar to those observed in younger mice. PARP1 levels were a similar story and PARP1 and DBC1 bonded proteins were reduced. The researchers also recorded a reduction in biomarkers of DNA damage suggesting that DNA repair had been improved.
Finally, the researchers exposed mice to radiation to damage their DNA. They discovered that mice treated with NMN before radiation exposure showed lower levels of DNA damage. The mice also did not display the characteristic changes to blood counts, such as changes to lymphocyte and hemoglobin levels, typically seen after radiation exposure. Interestingly, mice treated after radiation also enjoyed similar protective effects from NMN treatment.
Conclusion
Human trials with NMN are anticipated to begin within the next six months according to the researchers, and the potential discoveries are significant for our understanding of the biology of aging.
In closing, the results reveal a previously unknown mechanism behind DNA repair and cell death caused by DNA damage. Should further animal studies and human clinical results confirm the findings, this might pave the way for therapies that repair DNA damage due to radiation exposure from radiotherapy and the environment along with possibly treating age-related decline.
As always in science, we should remain optimistic, but further studies are needed before we can draw any final conclusions. We look forward to seeing the results of clinical trials in the near future.
Literature
[1] López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2013). The hallmarks of aging. Cell, 153(6), 1194-1217. [2] Schultz, M. B., & Sinclair, D. A. (2016). Why NAD+ Declines during Aging: It’s Destroyed. Cell metabolism, 23(6), 965-966. [3] Li, J., Bonkowski, M. S., Moniot, S., Zhang, D., Hubbard, B. P., Ling, A. J., … & Aravind, L. (2017). A conserved NAD+ binding pocket that regulates protein-protein interactions during aging. Science, 355(6331), 1312-1317. [4] North, B. J., Rosenberg, M. A., Jeganathan, K. B., Hafner, A. V., Michan, S., Dai, J., … & van Deursen, J. M. (2014). SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. The EMBO journal, e201386907. [5] Gomes, A. P., Price, N. L., Ling, A. J., Moslehi, J. J., Montgomery, M. K., Rajman, L., … & Mercken, E. M. (2013). Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell, 155(7), 1624-1638.