Cancer is the poster child of age-related diseases, and a recent study sheds light on why the risk of cancer rises dramatically as we age.
Abstract For many cancer types, incidence rises rapidly with age as an apparent power law, supporting the idea that cancer is caused by a gradual accumulation of genetic mutations. Similarly, the incidence of many infectious diseases strongly increases with age. Here, combining data from immunology and epidemiology, we show that many of these dramatic age-related increases in incidence can be modeled based on immune system decline, rather than mutation accumulation. In humans, the thymus atrophies from infancy, resulting in an exponential decline in T cell production with a half-life of ∼16 years, which we use as the basis for a minimal mathematical model of disease incidence. Our model outperforms the power law model with the same number of fitting parameters in describing cancer incidence data across a wide spectrum of different cancers, and provides excellent fits to infectious disease data. This framework provides mechanistic insight into cancer emergence, suggesting that age-related decline in T cell output is a major risk factor.
Thymic involution and rising cancer risk with age
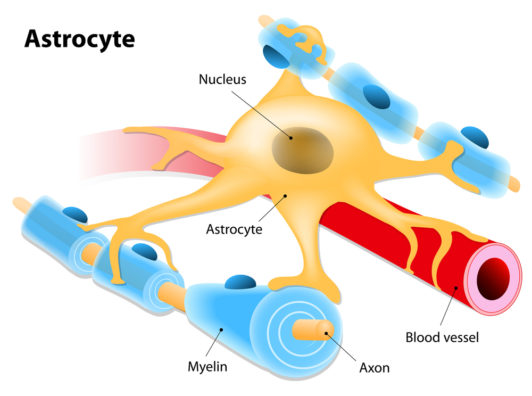
The thymus is a specialized organ that produces the majority of immune T cells, and as we age, it shrinks in size and ability to produce these cells. This process is called involution.
The involution of the thymus is part of the wider phenomenon of immunosenescence, the gradual deterioration of the immune system caused by aging. Immunosenescence involves the decline of the host’s ability to respond to infections and injury along with the immune system’s ability to remember previously encountered pathogens as part of immune memory.
Immunosenescence is observed in all species of animals that age regardless of lifespan, and it is relative to their life expectancy. It is a major factor in the increase in morbidity and mortality in older people. The decline of the immune system exposes a person to age-related and other diseases, particularly cancer.
A new model for cancer risk
It has been known for many decades that DNA mutations caused by genetic predisposition, environmental stressors, or lifestyle choices can cause cancer. The traditional view in science has been that cancer risk rises with age due to gradually increasing cellular mutations and that five or six mutations in a cell could cause it to turn cancerous.
However, earlier this year, a study by researchers at Heriot Watt University, the University of Edinburgh, and the Institut Curie in France suggests that immunosenescence could be the primary reason why cancer risk rises dramatically with age [1].
The team hypothesized that immunosenescence due to aging could result in a higher incidence of cancer, much as the incidence of other diseases is seen in older people. The researchers examined data from 2 million cancer cases of people in the 18-70 age range. Then, using a mathematical equation for how they expected cancer incidence to increase in relation to immunosenescence, they compared it to the age range profiles of 100 different kinds of cancer.
What they found was that their model was better supported by the data than the traditional mutation hypothesis for cancer. Their model also explains the difference in cancer incidence between men and women; the immune system tends to decline more slowly in women than it does in men. This is something that the traditional mutation hypothesis cannot really explain adequately.
Their findings suggest that it is the decline of the immune system during immunosenescence that plays a greater role in cancer development than mutations. This makes sense because cancer is constantly happening in the body, but the immune system patrols for, detects, and destroys it before it can spread.
Some of the most critical cancer-fighting immune cells are the T cells produced by the thymus. As aging causes the organ to shrink and turn into fat, the output of T cells falls to critically low levels and opens us up to cancer and other diseases.
If further studies confirm the validity of this new model of cancer prediction, it has significant implications for how we predict, prevent and treat cancer. This also means that the thymus is a prime target for regenerative medicine in the fight against age-related diseases.
We had the opportunity to speak with one of the study authors, Dr. Sam Palmer from Heriot-Watt University, about the experiment and his work as a researcher.
Some people suggest that cancer is the poster child of age-related diseases; given the results of your study, would you agree with the idea that cancer is primarily an age-related disease?
Absolutely, age is the single biggest predictor of cancer risk, and both cancer and aging itself have genetic mutations acting as a key player. I actually suspect that if something close to a cure for cancer is found, it will be intimately linked with pro-longevity treatments. So, curing cancer and curing aging may well go hand in hand.
Another poster child for age-related disease is Alzheimer’s disease. I actually checked to see if the risk of neurodegenerative diseases would fit the model, and their profiles are quite different. Risk comes on suddenly and rises exponentially at a very fast rate, doubling every 4 years or so. This is much faster than the relatively slow increase for cancer and infectious diseases, which often double every 16 years. As reported in my paper, this exponential increase in risk is mirrored by the exponential decay of the thymus (and T-cell production), which halve every 16 years.
As a researcher, what prompted you to develop the immunosenescence model for cancer?
This is quite a long story but, hopefully, an interesting one. It all started with a paper from Tomasetti and Vogelstein that made headlines for claiming that 2/3rds of cancer is “bad luck”. While I don’t agree with the bad luck interpretation, what they found was very interesting. They found a correlation between the number of stem cell divisions in a given organ and the risk of cancer in that organ.
So as I stewed over that, I noticed something strange. The traditional idea that cancer arises from a gradual accumulation of mutations (which I believed at the time) would actually predict a non-linear relationship, rather than what they found. So, I thought about why people believe this mutation accumulation idea, and one reason is that risk often goes up with age as a “power law”, as in, risk is proportional to age to some power. This is what you would expect from waiting for some rare events. These rare events were interpreted as driver mutations, and this line of reasoning is seen in textbooks, etc.
So, I looked into it myself and confirmed that some cancers do indeed rise as power laws, but I noticed some cancers, such as CML and brain cancers, rising exponentially, which is not what you would expect from accumulating mutations. So, I thought, what other factor might lead to an increase in risk with age? That’s when I started investigating the decline of the immune system and found that T-cell production declines exponentially at the same rate that those exponential cancers rose.
Then, I looked into infectious diseases and found many of them rising in risk exponentially, doubling every 16 years as well. So I thought about making a model to describe the interactions of T-cells and antigenic cells, that would capture this inverse correlation. After a few attempts that predicted different types of relationships, I arrived at a model, which I imagined as a kind of war between T-cells and cancer cells, which the cancer cells would win if they manage to grow up to a certain size. Then, surprisingly, with my group at the University of Dundee, we found that this model could also fit cancers like colon cancer, which happen to look like power laws. Overall, when fitting to all cancer types, this model fit slightly better than the power law model, with the same number of fitting parameters.
What do you think was lacking in the traditional mutational model for cancer?
Initially, nothing! It was only when I saw that certain cancers rise in risk exponentially, rather than as power laws, that I really started to doubt it.
For cancers that rise as power laws, the traditional mutational model for cancer, we would predict that the number of driver mutations is usually around six. However, there was a paper which took a different approach and found that “only three driver gene mutations are required for the development of lung and colorectal cancers”, and this seems quite solid to me. If that is right, then you can break down the rise in risk as partly coming from accumulating three driver mutations and partly from immune decline. If you do that, it turns out that immune decline is an even bigger factor than the mutation accumulation factor.
We actually made a combined model to include both factors, and this model also arrived at the estimate of three driver mutations. For the cancers that rise exponentially, this model suggests just one driver mutation. One such cancer is CML (chronic myeloid leukaemia), which already has a good candidate for what this one driver mutation would be: it is known as the Philadelphia chromosome and comes from a chromosomal translocation event.
Your study suggests that unlike the traditional view of cancer emergence in which multiple mutations are required, it appears that even single oncogenic mutations can lead to cancer if the immune system does not destroy the affected cells. Obviously, in an older person with a much less active immune system, that would mean a rising cancer risk. In your view, are most cancer cases a consequence of the immune deficiency caused by aging and resulting thymic involution?
That’s right. This is known as the immunosurveillance hypothesis, and there is some evidence for it.
In your paper, you note that the body does continue to create T cells even when the thymus has involuted to the point that it is effectively useless. T cells are produced elsewhere in the body, particularly via peripheral clonal expansion. However, despite the level of T cells remaining relatively constant even in old age due to clonal expansion, these T cells do not appear to do a very good job at keeping cancer at bay; why is this?
That’s right, although I’m not sure the thymus is ever effectively useless, but it does get very small. T-cells develop from hematopoietic stem cells; they first develop a “random” T-cell receptor then pass through a selection process in the thymus, ensuring that they are not self-reactive, then they replicate (clonal expansion). With age, the rate of T-cell production goes down, but the rate of clonal expansion goes up, resulting in an approximately constant total number of T-cells. This finding that T-cell production is inversely proportional to disease risk suggests that this clonal expansion does not increase the effectiveness of T-cells. I think testing that directly would make a great experiment.
There was an interesting paper recently that showed that the effectiveness of T-cells depends linearly on co-stimulatory factors. Perhaps whatever mechanism is behind that may explain both phenomena.
There are a number of research teams engaged in trying to encourage the thymus to regrow; are you optimistic that we can achieve rejuvenation of the thymus, and what impact do you think it might have, not only in terms of cancer but for other age-related diseases?
I am optimistic. One of the co-authors of this project, Prof. Clare Blackburn at the University of Edinburgh, has already succeeded in reprogramming fibroblast cells of mice to create a lab-grown mouse thymus. She is now working on doing the same for a human thymus. Other people are taking different approaches, and so it seems very likely that one of them will succeed soon.
As for what impact it might have, I can only speculate. As a best-case scenario, it may provide a treatment for various diseases, and perhaps the new T-cells may even clear out mutated (non-cancerous) cells, which would effectively treat aging itself. I find that idea really exciting because T-cells already have this amazing method for detecting mutated cells, so perhaps that could be used to kill dysfunctional cells and prolong lifespan. However, even if a T-cell recognizes a cell as mutated, if there are no “danger signals”, it won’t actually do anything. So I suspect that additional steps will be needed for thymus regeneration to delay aging itself.
There have been some small-scale experiments with mice, mostly from the 1970’s, looking at whether thymus transplantation affects lifespan or cancer progression. The experiments had very small sample sizes, so take them with a grain of salt. The lifespan extension results were not very promising, but the cancer experiments did appear to work. So, we’ll have to wait and see.
What is the next step in developing your immunosenescence model of cancer and how has it been accepted so far in academia?
There are several possible next steps. I think one obvious one would be to see if a thymus transplant could cure cancer in mice, essentially repeating the experiments from the 1970s with a larger sample size.
Another possible experiment would be to see if the effectiveness of T-cells is independent of clonal expansion. In particular, I would predict that there is an ‘immune escape threshold’ proportional to the number of T-cell clones, that doesn’t depend on how much clonal expansion the T-cells have done. Apart from that, there are lots of potential ideas: applying this framework to autoimmune diseases, childhood infections and cancers, cancers with quantifiable risk factors such as lung cancer, etc.
I think the response so far has been very positive. Some discussions followed that were published separately as letters, but, so far, no one has pointed out any fatal flaws. I think that this paper is part of a general shift in perspective, which is happening, where the immune system is playing a more central role. There was even a paper earlier this year which began with the very audacious opening line: “It has recently become apparent that the immune system can cure cancer.”
Thank you very much to Dr. Palmer for taking the time to speak to us about this fascinating study.
If you would like to read more about attempts to restore the thymus, you may find our interview with Dr. Greg Fahy last year to be interesting. Dr. Fahy has been developing a method to encourage the thymus to regrow, and, last year, he concluded a pilot study in a small group of people with some positive results.
Dr. Fahy has recently formed Intervene Immune, a biotech company focused on rejuvenation of the immune system.
Literature
[1] Palmer, S., Albergante, L., Blackburn, C. C., & Newman, T. J. (2018). Thymic involution and rising disease incidence with age. Proceedings of the National Academy of Sciences, 115(8), 1883-1888.
[2] Bredenkamp, N., Ulyanchenko, S., O’Neill, K. E., Manley, N. R., Vaidya, H. J., & Blackburn, C. C. (2014). An organized and functional thymus generated from FOXN1-reprogrammed fibroblasts. Nature cell biology, 16(9), 902.