Welcome to part two of our three-part Undoing Aging 2018 interview of Dr. Aubrey de Grey and his team at SENS Research Foundation. Today, we have some of the scientific questions that the community had about SENS; there are some very detailed responses, and we hope you enjoy them.
Regarding the use of senolytics, are you concerned about their potential to remove highly specialized cells like cardiomyocytes, which do not divide or do so very slowly? Could taking senolytics without the ability to replace these specialized lost cells be risky unless combined with replacement therapies?
Aubrey: This is not a major concern, for a few reasons. First, when cells turn senescent, they cease carrying out their specialized function (as a cardiomyocyte, or neuron, or what have you), so no such function is lost by ablating them. Second, cells that don’t divide (like cardiomyocytes and neurons) are far less likely to become senescent in the first place than cell types that divide; many of the main drivers of senescence are related to cell division. And third, in the specific case of cardiomyocytes, there’s already significant evidence in rodents that it improves cardiac function overall [1] as well as wider cardiovascular health [2-3].
However, there is some reason for concern here, which is why we’re already working to develop the next generation of senescent cell ablation therapies. The selectivity of senolytic drugs for senescent cells comes from the fact that they target the activity or expression of genes involved in cell survival, on which senescent cells are much more reliant than healthy cells under normal, unstressed conditions. But during times in which the cell is under stress, normal cells also rely on those same pathways to carry them through and give them time to recover. Thus, although the net effect of these drugs is undeniably positive, their mechanism of action will necessarily entail occasionally killing off healthy cells that are experiencing a moment of vulnerability when the drug is administered and that they could otherwise have survived. Again, such cells could include difficult-to-replace cells like heart muscle cells and neurons.
Future therapies can target truly senescent cells more selectively, and SENS Research Foundation is helping to advance those next-generation senescent cell therapies even as UNITY prepares for human testing through our investment in Oisín Biotechnologies. We’ll have more to say on this in an upcoming blog post.
But. certainly, no single rejuvenation biotechnology will work as well on its own as it will as part of a comprehensive panel of such biotechnologies, and matching senescent cell ablation with cell replacement therapies is one of the most straightforward examples.
Senolytic drugs gave mice about 35% increased healthy lifespan in experiments. Given that every living organism produces senescent cells the same way, could this mean that it may translate to humans?
Aubrey: First, let’s be clear on just what senescent cell ablation has been shown to do. In the study of which you’re thinking [4], senescent cell ablation didn’t extend lifespans by 35%; it increased median lifespan by 24-27% under most conditions, with no effect on maximum lifespan. It only increased median lifespan by 35% in a subset of animals where the controls were exceptionally short-lived compared to all the other animals in the study. (In fact, all the animals in the study were at least a bit shorter-lived than healthy mice normally are, probably due to some combination of the stress of twice-weekly injections and possibly some effect of the transgenes, though the latter is probably minor — but the 35% figure is clearly not robust and certainly shouldn’t be extrapolated to normal, otherwise-healthy aging humans).
Now, confining ourselves to that 24-27% median lifespan figure: interventions that lead to gains in median lifespan only in laboratory mice, with no corresponding effect on a robust maximum lifespan (tenth-decile survivorship), still need to be heavily discounted when speculating on effects in humans. Interventions that only affect median lifespan primarily affect deaths in the first half of the lifespan — and here there is a critical difference between mice in a lab and modern humans, for whom medicine has already eliminated many causes of such early deaths, from vaccines (which also impact late-life mortality by reducing lifelong inflammatory burden), to surgery, to antibiotics, to drugs that more obviously affect middle-aged people.
The force of this reasoning is somewhat attenuated in the case of interventions like senescent cell clearance, which actually repair aging damage, than with interventions affecting environmental or metabolic risk factors driving “premature” disease (obesity, inflammation, cardiovascular risk factors, environmental toxins, etc). Still, you have to assume that the effect on lifespan of any single damage-repair intervention in isolation will be modest, based on the principle of the “weakest link in the chain”: all the links are weakening over time, and shoring up only one of them still leaves the rest of the links damaged and ready to shear, whereupon the whole chain is broken. To move the needle on lifespan in modern humans, we have to push back on all of the cellular and molecular damage of aging, not just one form.
Are senescent cells fewer in number or less destructive in humans than in other animals because we are more advanced organisms than they are?
Aubrey: Evolutionary biologists would quibble with the notion that humans are “more advanced” organisms than mice (or even than roundworms), but we’re certainly longer-lived organisms than they are — and of necessity, this entails that the rate of accumulation of all the cellular and molecular damage of aging — including senescent cells — is slower in us than in them.
Some proteins can facilitate DNA repair. However, what can SENS do to prevent cells from collecting DNA damage in addition to the proteins that already exist in our body? Also, what approaches are being taken to correct benign mutations?
Aubrey: Because the levels or activity of some of the proteins that repair DNA are downregulated by the downstream metabolic effects of aging damage (such as inflammation and oxidative stress), or are known to be downregulated with age for unknown reasons, removing the underlying damage driving these age-related declines (such as by ablating senescent cells and rendering mitochondrial mutations harmless) will “take the brakes off” these proteins and restore their ability to repair DNA to the youthful norm, just as rejuvenation biotechnology will reset other downstream derangements of the aging metabolism.
By “benign mutations,” I take it that you mean mutations that don’t cause cancer. Regardless, there is no foreseeable technology (meaning, a technology that can be described in detail and is technically feasible to implement within the next 2-3 decades) that will be able to correct existing mutations. Instead, the focus must be on removing, repairing, or obviating the effects of mutations that are relevant to our health over the course of currently-normal lifespans: clearing senescent cells, replacing cells lost to apoptosis and senescence and other causes, and making the body impervious to cancer. That will buy us time during which scientists can develop future generations of rejuvenation biotechnology to repair DNA mutations directly.
Regarding the breakdown of extracellular aggregates, what will you do if the first wave of treatments using antibodies is unable to repair the whole system?
Aubrey: Certainly, it’s guaranteed that no first-generation SENS therapy will be able to repair every single contributor to any given category of aging damage — and it doesn’t have to. All we have to do to reach “longevity escape velocity“ is to remove or repair the specific forms of cellular and molecular aging damage within each category that meaningfully restrict our lives to the extremes of current lifespans. During the extra decades of healthy life that we’ll then enjoy, scientists can then work to identify the constraints that limit life- and healthspan to those newly-expanded horizons.
Accordingly, all SENS therapies will need to be iteratively improved; we will want safer and more effective ways to repair the damage targeted by earlier iterations of rejuvenation biotechnologies and also to repair additional specific targets within each category. It’s only once those first therapies are developed and in use that we’ll know what their specific limitations will be; what the relative prioritization and, in most cases, even the identities of the next-most important targets will be (our project on Target Prioritization of Adventitious Tissue Crosslinking is tackling a notable exception); and how exactly to design improved or new therapies in each category.
In old age, the vitreous body—an acellular component of the eye—liquifies. The resulting change in viscosity may cause a post-vitreous detachment that can be dangerous for the retina. Where does the relevant age-related damage fit within the SENS categorization? What regenerative interventions might be applied to reverse it?
Aubrey: As with all age-related degeneration, post-vitreous detachment (PVD) can only ultimately flow from stable changes in the cellular and molecular structures responsible for normal, youthful vitreous function. However, the structural basis for PVD is unfortunately still poorly understood. Up until recently, it’s been difficult to study the organ at all, let alone its aging, because of the inherent difficulty of visualizing a tissue that is, by design, invisible and because many of the techniques that have historically been used to study it have required the use of reagents that precipitate the jelly-like material out of the vitreous humor [5]. And even today, with better tools available, there is precious little research in this area.
But let’s focus on what we do know. The vitreous is composed of a network of collagen fibres that are coated by non-covalently bound structural molecules (glycoproteins and chondroitin sulfate) that allow the collagen fibers to slide past one another without sticking to each other and also to interact with the gel phase of the vitreous (primarily comprised of a glycoprotein called hyaluronan). In youth, most of the hyaluronan remains in a gel phase, but over the lifespan, a rising amount of it is degraded into a liquid phase. This degenerative process is already apparent in four-year-old children, and liquid-phase vitreous occupies about 20% of the total vitreous volume by the time one is in one’s late teens; this process accelerates after age 40, to the point where more than half the vitreous has been degraded into a liquid in octogenarians. Along with the shift from gel phase to liquid phase, there is a reduction in the volume of gel vitreous, without a change in total collagen until the extremes of current lifetimes [5].
As the gel shrinks, it begins to separate from the retina, with the gap filled by the accumulated liquid vitreous. If this process of separation happens too quickly in a given area, or if the vitreous gel and the retina remain adherent despite the contraction of the vitreous, then the retina or a retinal blood vessel can tear, leading to symptoms like “flashes” and “floaters.”
As of yet, we don’t know what’s driving these processes. The most suspicious change in the structure of the vitreous with age is the “lateral aggregation” of the collagen fibrils — in other words, the bunching-together of adjacent collagen fibrils. The two prime suspects for this aggregation are age-related loss of the coating proteins that keep the collagen fibrils from naturally sticking to their neighbors, and AGE or other crosslinks forming between the fibrils.
Abnormal AGE crosslinks certainly do occur in the collagen in the vitreous of diabetics, which likely contributes to diabetic retinopathy, an important complication in diabetes that leads to blindness [5]. To the extent that those are involved in PVD in aging nondiabetics, AGE-breakers could be brought to bear to liberate the bound collagen fibrils, allowing them to support gel-phase vitreous hyaluronan again.
Alternatively, the aggregation may result from an age-related loss of the coating proteins that keep the fibrils from sticking together [6]. There are several protein-degrading enzymes in the vitreous that could, in principle, do this if their expression rises with age, and aggregation and liquefaction can be triggered in the lab by injecting any of several common physiologic enzymes into the vitreous. Conversely, peptides have been designed that shield the coating proteins from some of these enzymes; these peptides have protective effects against degradation of bovine vitreous treated with some suspect enzymes, but not others [7].
Along with trypsin, matrix metalloproteinases (MMPs) are among the enzymes most strongly suspected of involvement in an age-related rise in such “stripping” of vitreous collagen [6]. While the link has not been directly made, several lines of circumstantial evidence suggest that a rise in MMP levels with age could be driven by the accumulation of senescent cells in the eye. Senescent cells do accumulate in the retina and other ocular tissues with age, and MMPs are a component of the toxic soup that senescent cells secrete — the so-called senescence-associated secretory phenotype or SASP. And while they didn’t look specifically at MMPs, one study did find increased levels of other components of the SASP in the vitreous of patients suffering from proliferative diabetic retinopathy [8]. If senescent cells are indeed driving an age-related rise in collagen-stripping MMPs, then ablating those cells would put a stop to it, potentially preventing or reversing PVD.
Another possible contributor to PVD is damage to hyaluronan molecules by free radicals, which warp the three-dimensional structure of hyaluronan in model systems. Lifelong exposure to free radicals from metabolic processes and/or ultraviolet light could cause structural changes that either cause gel-phase hyaluronan to dissociate from collagen fibrils or damage hyaluronan decorating adjacent collagen fibrils so that they no longer slide past each other but instead aggregate, leading to liquefaction and PVD [5]. Rendering mitochondrial mutations harmless would eliminate the main driver of the age-related rise in oxidative stress.
Again, none of this is certain; experts don’t know for sure what’s driving the damage underlying PVD, and we’ll need to understand that in order to know what rejuvenation biotechnologies will prevent and treat it. But as with all diseases and disorders of aging, structural changes are driving it, and structural remediation will be the key to ending it; looking at the existing lineup of suspects, it appears that all can be addressed with therapies that contribute to planks of the existing SENS platform.
Increased anabolic signalling, which signals an abundance of nutrients, appears to accelerate aging, while decreased anabolic signalling is shown to extend lifespan. Does this suggest that excessive caloric intake accelerates aging and that a reduced intake may slow it down? If so, may practices such as bodybuilding, which require significant food intake, lead to accelerated aging?
Aubrey: Anabolic signalling is one important driver of the cellular and molecular damage that accumulates over a lifetime, leading ultimately to age-related pathology. This is even true of the normative physiological level of anabolism that supports processes like normal growth and development; wound healing and other regenerative responses; and maintenance of a lean adult body plan. And many interventions that decrease anabolic signalling below this physiologic level meaningfully slow aging in rodents and other relatively short-lived animals; reducing energy intake (i.e., calorie restriction (CR)) and mutations in insulin-like growth factor-1 (IGF-1) pathway are well-known examples.
However, even in rodents, it’s not clear that increasing anabolic signalling above the normative physiological level hastens aging relative to the base case. Obesity, of course, is bad for your health, whether you’re a (wo)man or a mouse — but it’s not clear that the reason it’s harmful is just a matter of “more of the same” anabolic stimuli that contribute to aging in the base case, rather than primarily a distinct pathophysiological process. And the ill-health of obesity is clearly not the simple inverse of the slow-aging phenomenon of low-anabolic states like CR. Further complicating matters, it’s important to bear in mind that there is significant debate as to whether the age-retarding effects of low-anabolic states like CR meaningfully impact aging in humans or other long-lived species [9-10].
Of course, bodybuilders are both lean and have high energy intake. This is also true of endurance athletes, and it’s clear in both rodents and humans that endurance exercise is healthy and does not appear to either accelerate or decelerate aging. The data are significantly more sparse as regards the specific effects of bodybuilding. It’s a difficult kind of activity to model in rodents, and it’s also more difficult to study in the long term in free-living humans than endurance exercises like running: fewer people bodybuild, they are less likely to carry on with it past middle age, there is greater variation in training routines, and the data are confounded by the prevalent abuse of anabolic steroids and other drugs, even among amateurs.
Whatever its effects on “aging itself,” there’s certainly compelling evidence that modest levels of strength training are good for your long-term health, associated with similar or lower risk of mortality as compared with endurance exercise [11], although elite bodybuilders may lose some of its benefits [12-14]. A sensible approach would be to pursue strength training without seeking to push the limits of your genetic potential through very high energy and protein intake or the use of anabolic steroids or IGF-1; this will improve your insulin sensitivity and reduce your risk of osteoporosis and premature frailty, with less risk of injury or truly harmful levels of anabolic signaling.
Has SENS/Aubrey reviewed their position that nuclear mutations matter only in cancer in light of recent research results suggesting that certain ominous mutations in hematopoietic stem cells increase the risk of developing not only blood cancers (50 fold) but dying of all causes (by 40%), particularly cardiovascular diseases, including atherosclerosis and stroke?
Aubrey: The research on this “clonal hematopoiesis” phenomenon is certainly provocative but doesn’t ultimately change our view on this question. Remember first that it has never been our position that nuclear mutations matter only in causing cancer; at a minimum, they also matter in causing apoptosis (“cellular suicide,” which denudes the body of functional cells with age, most importantly stem cells) and cellular senescence (ditto, plus the baleful effects of the SASP). And then remember that SENS is fundamentally an engineering approach to aging, focused on practical solutions rather than acquiring a full understanding of mechanistic details. Our position has been, therefore, that all the effects of nuclear mutations that meaningfully constrain current human lifespan/healthspan can be obviated by removing, repairing, or obviating the effects of mutations that are relevant to our health over the course of currently-normal lifespans: clearing senescent cells, replacing cells lost to apoptosis and senescence and other causes, and making the body impervious to cancer.
In clonal hematopoiesis, blood stem cells with one of a small number of mutations gain a selective advantage over blood stem cells with other genotypes, which allows them to “take over” the stem cell compartment. [15] This isn’t exactly what an oncologist would call “cancer,” but it is a clear case of “cells too many” caused by nuclear mutations proliferating at the expense of their neighbors, which fits the operational criteria for the oncoSENS category. And the periodic purging of all native bone marrow stem cells and their wholesale replacement with fresh, mutation-free, cancer-proof ones — which would immediately eliminate clonal hematopoiesis — is already planned to be the very first clinical phase of the WILT plan to pre-emptively shut down cancer.
Even before we begin implementing WILT, there are rejuvenation biotechnologies in the SENS platform that can minimize the harms that these aberrant cells are suspected of causing. In reported studies, the cause of the excess non-cancer mortality associated with clonal hematopoiesis has been death from cardiovascular disease and stroke [16-18]. In an accompanying animal study, the investigators showed that this could be accounted for by changes in the macrophages derived from bone marrow-bearing clonal hematopoiesis mutations; these macrophages express higher levels of inflammatory mediators that contribute to atherosclerosis than macrophages derived from normal bone marrow [9]. Work by independent researchers also finds that the gene whose loss is modelled in that study is essential to the differentiation of macrophages [19], which could be an additional mechanism.
Of course, atherosclerosis and stroke can be prevented by lysoSENS rejuvenation biotechnology: clearing the macrophage/foam cell lysosome of cholesterol waste products, ablating senescent arterial macrophages and smooth muscle cells, [2, 3] and to a lesser extent, reversing large artery stiffness. So, again, we have ways to deal with the harms that clonal hematopoiesis causes, despite having no medium-term prospects for reversing the underlying mutations.
Finally, it should also be emphasized that this phenomenon should not be extrapolated to other aging tissues. Clonal hematopoiesis is enabled by the very high genetic diversity of the blood stem cell compartment and its high rate of replication as compared to other tissues (even other stem cell compartments) — and even with those enabling characteristics, only about 10%–20% of people develop it by the time they are in their 70s [16]. No other non-cancerous cell types have this inherent potential for mutation-driven clonal expansion.
And while anecdotal, we should also note the case of a female supercentenarian whose exceptional longevity (by current, unremediated aging standards) was still possible despite having nearly all of her blood stem cell compartment dominated by two such clonal lines [19].
That concludes part two of our Undoing Aging 2018 interview; we’ll publish the third and final part tomorrow here on our blog. If you missed part one you can find it here.
Literature
[1] Zhu, Y., Tchkonia, T., Pirtskhalava, T., Gower, A. C., Ding, H., Giorgadze, N., … & O’hara, S. P. (2015). The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging cell, 14(4), 644-658.
[2] Roos, Carolyn M., Bin Zhang, Allyson K. Palmer, Mikolaj B. Ogrodnik, Tamar Pirtskhalava, Nassir M. Thalji, Michael Hagler et al. “Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice.” Aging Cell 15, no. 5 (2016): 973-977.
[3] Childs, B. G., Baker, D. J., Wijshake, T., Conover, C. A., Campisi, J., & Van Deursen, J. M. (2016). Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science, 354(6311), 472-477.
[4] Baker, D. J., Childs, B. G., Durik, M., Wijers, M. E., Sieben, C. J., Zhong, J., … & Khazaie, K. (2016). Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature, 530(7589), 184.
[5] Sebag, J. (1992). Anatomy and pathology of the vitreo-retinal interface. Eye, 6(6), 541.
[6] Bishop, P. N., Holmes, D. F., Kadler, K. E., McLeod, D., & Bos, K. J. (2004). Age-related changes on the surface of vitreous collagen fibrils. Investigative ophthalmology & visual science, 45(4), 1041-1046.
[7] Zhang, Q., Filas, B. A., Roth, R., Heuser, J., Ma, N., Sharma, S., … & Shui, Y. B. (2014). Preservation of the structure of enzymatically-degraded bovine vitreous using synthetic proteoglycan mimics. Investigative ophthalmology & visual science, 55(12), 8153-8162.
[8] Oubaha, M., Miloudi, K., Dejda, A., Guber, V., Mawambo, G., Germain, M. A., … & Mallette, F. A. (2016). Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Science translational medicine, 8(362), 362ra144-362ra144.
[9] de Grey, A. D. (2005). The unfortunate influence of the weather on the rate of ageing: why human caloric restriction or its emulation may only extend life expectancy by 2–3 years. Gerontology, 51(2), 73-82.
[10] Rae, M. J. (2006). You don’t need a weatherman: famines, evolution, and intervention into aging. Age, 28(1), 93-109.
[11] Stamatakis, E., Lee, I. M., Bennie, J., Freeston, J., Hamer, M., O’Donovan, G., … & Mavros, Y. (2017). Does strength promoting exercise confer unique health benefits? A pooled analysis of eleven population cohorts with all-cause, cancer, and cardiovascular mortality endpoints. American journal of epidemiology.
[12] Clarke, P. M., Walter, S. J., Hayen, A., Mallon, W. J., Heijmans, J., & Studdert, D. M. (2015). Survival of the fittest: retrospective cohort study of the longevity of Olympic medallists in the modern era. Br J Sports Med, 49(13), 898-902.
[13] Teramoto, M., & Bungum, T. J. (2010). Mortality and longevity of elite athletes. Journal of Science and Medicine in Sport, 13(4), 410-416.
[14] Sarna, S. E. P. P. O., Sahi, T., Koskenvuo, M. A. R. K. K. U., & Kaprio, J. A. A. K. K. O. (1993). Increased life expectancy of world class male athletes. Medicine and science in sports and exercise, 25(2), 237-244.
[15] Jan, M., Ebert, B. L., & Jaiswal, S. (2017, January). Clonal hematopoiesis. In Seminars in hematology (Vol. 54, No. 1, pp. 43-50).
[16] Jaiswal, S., Natarajan, P., Silver, A. J., Gibson, C. J., Bick, A. G., Shvartz, E., … & Baber, U. (2017). Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. New England Journal of Medicine, 377(2), 111-121.
[17] Jaiswal, S., Fontanillas, P., Flannick, J., Manning, A., Grauman, P. V., Mar, B. G., … & Higgins, J. M. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. New England Journal of Medicine, 371(26), 2488-2498.
[18] Kallin, E. M., Rodríguez-Ubreva, J., Christensen, J., Cimmino, L., Aifantis, I., Helin, K., … & Graf, T. (2012). Tet2 facilitates the derepression of myeloid target genes during CEBPα-induced transdifferentiation of pre-B cells. Molecular cell, 48(2), 266-276.
[19] Holstege, H., Pfeiffer, W., Sie, D., Hulsman, M., Nicholas, T. J., Lee, C. C., … & Meijers-Heijboer, H. (2014). Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome research, 24(5), 733-742.


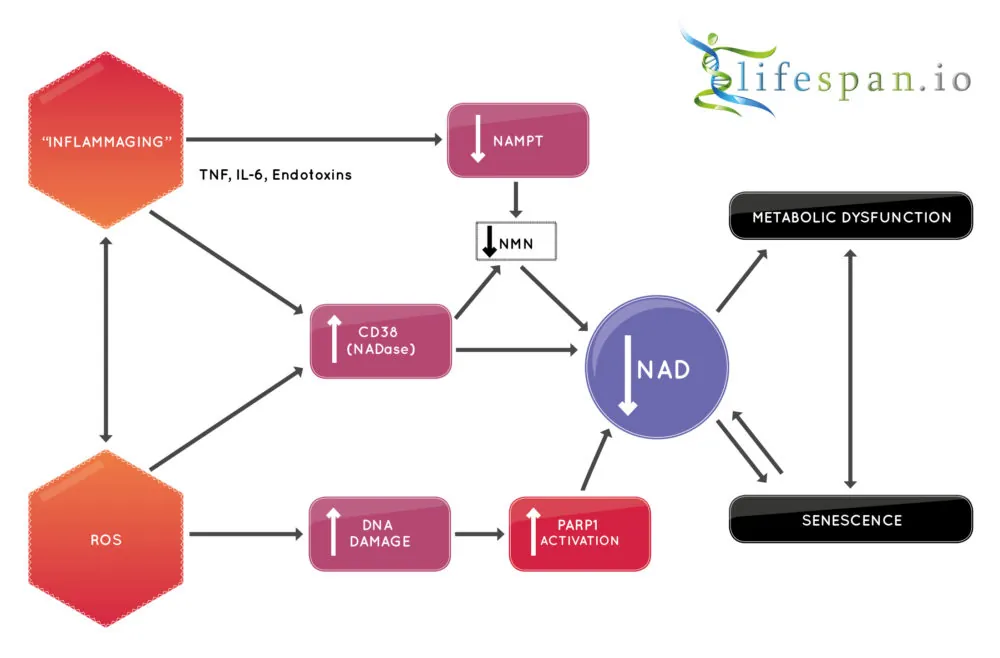 Therapies that increase NAD+ in cells, and potentially co-therapies that reduce CD38, are a potential way to slow an aging process and may help to treat metabolic disorders such as diabetes. There is potential for such approaches to increase the number of years we spend healthy, and it may even increase lifespan as it does in other species; the good news is that we may soon find out.
Therapies that increase NAD+ in cells, and potentially co-therapies that reduce CD38, are a potential way to slow an aging process and may help to treat metabolic disorders such as diabetes. There is potential for such approaches to increase the number of years we spend healthy, and it may even increase lifespan as it does in other species; the good news is that we may soon find out.