Chronic Inflammation
;){kind=link}
;){kind=link}
Chronic inflammation refers to a persistent, low-grade buzz of immune activity that settles into the body without the drama of an infection or obvious injury. In the world of aging, this slow, smoldering fire is often called inflammaging, a term coined by Claudio Franceschi to capture the systemic, hard-to-detect inflammation that creeps in with advancing years [1]. Acute inflammation is useful, representing an emergency medical team rushing to the scene. Chronic inflammation, on the other hand, is a fire alarm that won’t shut off even after the flames are long gone. Left unchecked, this background noise quietly erodes tissues, raising havoc one cell at a time.
Researchers have noted that as we age, the blood becomes a little like a suspiciously overcrowded chat room as it becomes filled with inflammatory signals. Pro-inflammatory cytokines like interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and C-reactive protein (CRP) start appearing in higher amounts, even in people who otherwise seem perfectly healthy. Elevated IL-6 levels, in particular, have become so reliable that they predict disability and even all-cause mortality among the elderly [2].
This quiet immune activity is not without consequence. Arteries thicken and harden [3], neurons lose their finely tuned communication [4], joints surrender to osteoarthritis, and spinal discs decay like old leather.
For a long time, chronic inflammation was considered more of a tag-along issue, lumped under broad changes in intercellular communication in the original Hallmarks of Aging paper by López-Otín and his team [5]. However, over the past decade, research has sharpened this view: chronic inflammation is a lead actor, not a background detail. It meets every criterion to be a true hallmark of aging: it increases with age, it accelerates aging and disease in experiments, and reducing it can extend healthspan and even lifespan [6].
In 2023, López-Otín and colleagues formally promoted chronic inflammation to its rightful status, naming it one of twelve key hallmarks of aging. The updated list includes the original troublemakers, such as genomic instability, telomere attrition, and epigenetic alterations, and includes three newly emphasized players: disabled macroautophagy, gut microbiome dysbiosis, and chronic inflammation [6].
Inflammaging’s rise to prominence wasn’t a courtesy title; it reflects the fact that chronic inflammation is both an outcome of other aging processes and an accelerant that fans their flames.
NF-κB activation and pro-inflammatory signaling
If chronic inflammation had a ringleader, it would be nuclear factor kappa B (NF-κB). This master transcription factor pulls the strings behind much of the body’s pro-inflammatory behavior. In a young, healthy body, NF-κB is briefly called into action when there’s trouble and steps back when the trouble subsides [7]. However, with age, the system frays. NF-κB stays switched on longer and more often, flooding tissues with inflammatory cytokines like IL-6, IL-1β, and TNF-α [8].
Aging cells offer no shortage of reasons to keep NF-κB active. DNA damage builds up over time, and the body’s damage sensors, like the ATM kinase and the cGAS-STING pathway, react by turning up the inflammatory volume [9]. What was once a well-timed emergency response turns into a perpetual background alarm. Persistent NF-κB activation doesn’t just keep cytokines flowing, it accelerates cellular aging itself. It encourages telomere shortening in critical stem cells, like those that regenerate muscle and liver tissue [10], and it promotes the genetic programs that lock cells into a non-dividing, senescent state [11].
To make matters worse, NF-κB also upregulates the production of enzymes, such as NOX-2, that churn out reactive oxygen species (ROS), compounding cellular damage. Instead of putting out fires, aging NF-κB turns into an arsonist, lighting small blazes in every corner [12].
One tangible consequence of this chronic activation is an excess of inflammatory cytokines spilling into the bloodstream. While IL-6 and IL-1β levels reliably rise with age, TNF-α levels show a more variable increase, particularly in older adults with metabolic or inflammatory conditions [12]. When elevated, TNF-α binds to its receptors and activates ROS-producing complexes like NADPH oxidase, amplifying oxidative stress. Meanwhile, IL-6 and IL-1β continue to fan the flames, driving systemic inflammation, insulin resistance, and muscle breakdown [13].
The entire cellular environment shifts toward a pro-inflammatory default. Animal studies have found that dialing down NF-κB activity in aged tissues can reverse some signs of aging. In mouse models of Alzheimer’s-like tau pathology, blocking NF-κB in the brain’s immune cells (microglia) reduced the spread of toxic tau proteins and improved cellular cleanup through autophagy. Conversely, cranking up NF-κB worsened the disease [14].
In the aging body, NF-κB isn’t merely a signal gone awry. It’s a perpetual siren that drowns out normal regulation, linking the everyday stress of being alive to the quiet, relentless march of inflammatory damage [15].
The NLRP3 Inflammasome and sterile inflammation
Another heavy hitter in the world of inflammaging is the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome. If cells are houses, inflammasomes are burglar alarms. Specifically, the NLRP3 inflammasome is the nervous neighbor who calls the police at the slightest whiff of trouble, and with aging, everything starts to look like trouble.
NLRP3 responds to a dizzying array of stress signals and cellular debris: ATP leaking out of damaged cells, bits of busted mitochondria, misfolded proteins, excess nutrients, even random metabolic leftovers. As we age, tissues accumulate more of these danger-associated molecular patterns (DAMPs), constantly priming and setting off NLRP3 [16].
One major offender is mitochondrial dysfunction. When old mitochondria start leaking their DNA and spewing reactive oxygen species into the cytosol, NLRP3 picks up the signal like a dog hearing a high-pitched whistle. Once activated, NLRP3 assembles a protein complex that flips the switch on caspase-1, an enzyme that processes pro-inflammatory cytokines, specifically IL-1β and IL-18, into their active, havoc-wreaking forms. Both cytokines are potent inflammatory agents, and studies have shown that IL-18 levels climb with age, even in otherwise healthy humans [17].
Crucially, NLRP3 doesn’t need an infection to cause trouble. It can trigger sterile inflammation, which means that the body starts an immune response against its own accumulating molecular junk. Essentially, the aged body becomes a battlefield littered with false alarms. Research shows that NLRP3 activity ramps up steadily with age [18]. Mice lacking NLRP3 show striking benefits: lower inflammation, stronger bones, better muscles, improved insulin sensitivity, sharper cognition, and a maximum lifespan nearly 30% longer compared to their normal, inflammasome-burdened peers. These NLRP3-deficient mice aged with far fewer of the typical degenerative changes [19, 20].
NLRP3 overactivation isn’t picky about what it damages. It’s been linked to atherosclerotic plaques clogging blood vessels [21], neurodegenerative disease in the brain [22], and inflammatory wear-and-tear in kidneys and hearts [23]. Mechanistically, NLRP3 sets up a vicious loop: the IL-1β and IL-18 it produces summon more inflammatory immune cells, and a particularly messy, inflammatory form of cell death known as caspase-1-driven pyroptosis dumps even more DAMPs into the surrounding tissues [14].
In short, NLRP3 turns a few broken mitochondria into a full-blown cellular neighborhood riot, which is why scientists are putting serious effort into figuring out how to disarm this particular alarm system before it burns the whole house down.
In summary, the NLRP3 inflammasome emerges as a critical driver of age-related sterile inflammation. With advancing age, its persistent activation leads to a steady drumbeat of IL-1β and IL-18 signaling, chipping away at tissue health over time. This makes NLRP3 one of the prime suspects for anyone interested in stamping out inflammaging. Suppressing NLRP3 has already shown promise in animal studies, where it dials down chronic inflammation and promotes healthier, longer-lived mice.
Senescent cells and the SASP
Another steady source of inflammaging lies in the buildup of senescent cells. Senescence is the biological equivalent of retiring workers after a lifetime of service but then letting them hang around the office, where they spread bad habits and negativity. When cells experience irreparable damage from telomere shortening, DNA breaks, or oncogene activation, they permanently exit the cell cycle. They stop dividing but don’t quietly disappear. Instead, they become active secretory units, pumping out a disruptive cocktail known as the senescence-associated secretory phenotype (SASP) [24].
The SASP is a raucous brew of cytokines like IL-6, IL-8, and TNF-α along with chemokines, proteases, and growth factors [25]. It’s largely orchestrated by NF-κB and stress pathways like p38 MAPK and C/EBPβ [26]. Essentially, these cells turn into miniature inflammation factories, continuously sending out distress signals into their local environment.
The influence of a single senescent cell can ripple outward. SASP factors draw in immune cells to the scene, which sometimes leads to clearance of the senescent cells but often just amplifies inflammation [26]. Even worse, these secreted factors can nudge neighboring healthy cells into a senescent, pro-inflammatory state themselves. It’s a biological version of a bad mood spreading through a crowded room [27].
As aging progresses, tissues gradually accumulate these inflammatory senescent cells. Blood biomarkers like matrix metalloproteinases and GDF15 climb higher in older adults, reflecting the systemic flood of SASP molecules [28].
The consequences aren’t just theoretical. Senescent cells actively drive pathology in aging. In atherosclerosis, they help build and destabilize plaques [29]. In osteoarthritis, they contribute to joint degeneration [30]. Encouragingly, senolytic drugs, which selectively kill senescent cells, have demonstrated that removing this toxic population can lower systemic inflammation and rejuvenate organ function in aged animals [31].
In short, the longer that senescent cells stick around, the louder and more destructive the SASP chorus becomes. Targeting these cells, or muting their inflammatory output, is now a major research frontier in the fight against inflammaging.
Immunosenescence and inflammaging
The degenerative processes of aging also manage to break the immune system in two opposite ways at once. On one hand, there’s immunosenescence, the slow crumbling of the immune defense machinery. On the other hand, there’s inflammaging, the background hum of pointless, chronic inflammation that refuses to die down [32].
In older adults, the immune system often sits in a state of low-level siege. Blood profiles show an uptick in pro-inflammatory immune cells and higher circulating levels of cytokines like IL-6, TNF-α, and interferons [33, 34]. Meanwhile, paradoxically, the system’s ability to mount a sharp, effective response to new threats is dulled. Vaccine responses weaken. Infections linger longer. It’s like having a security system that triggers a full SWAT team when a cat sneezes but fails to notice a burglar coming through the window.
Several changes contribute to this dysfunctional state. On the innate side, macrophages and neutrophils, which are normally the first responders, become trigger-happy and cranky. They spew reactive oxygen species and inflammatory cytokines even when nothing’s happening [33]. Meanwhile, pattern-recognition receptors like Toll-like receptors and inflammasomes such as NLRP3 become oversensitive, responding to harmless debris like it’s a five-alarm fire.
The adaptive immune system doesn’t fare much better. There’s a steady decline in naive T and B cells, rookies that are needed to deal with new infections, due to the shrinking of the thymus and changes in bone marrow. At the same time, the memory T cell pool fills up with exhausted, battle-worn veterans who secrete inflammatory mediators instead of contributing to useful defense [32]. Age-associated T cells are particularly dangerous CD4⁺ cells so battered by lifelong exposure to infections and autoantigens that they now specialize in spitting out granzyme K and wrecking nearby tissues [35, 36].
Old B cells also lose their edge. They become less diverse and skew toward producing autoantibodies, which help sustain the body’s slow internal fire [33].
The result is an immune system that both overreacts and underperforms. Chronic low-level immune activation damages tissues, while real threats slip through the cracks [33]. This misfiring immune landscape explains why elderly people are so prone to hyperinflammatory reactions during infections, such as the cytokine storms seen in some severe cases of influenza or COVID-19 [37] .
In short, aging delivers a brutal one-two punch to the immune system, making it inflamed, exhausted, and poorly regulated. The very system meant to protect us ends up fueling the flames of aging itself. Finding ways to recalibrate immune function, whether by clearing dysfunctional cells or rebalancing inflammatory responses, may be one of the most critical strategies for curbing inflammaging.
Systemic effects
Chronic inflammation doesn’t stay politely confined to a few misbehaving cells. It spreads, creeping through tissues and blood vessels like smoke leaking through a cracked door. Unlike acute inflammation, which shows up, handles business, and leaves, this slow-burn inflammation sticks around, quietly stressing every organ system it touches. Over time, the cumulative damage accelerates tissue degeneration, disrupts function, and writes aging into every cell’s ledger.
The cardiovascular system is one of the earliest victims. Blood vessels and the heart turn out to be particularly bad places to let a low-grade fire smolder. In aging arteries, inflammaging fuels the process of atherosclerosis, the hardening and narrowing of the arterial highways. Cytokines like IL-1β, IL-6, and TNF-α encourage endothelial cells, which make up the smooth lining of blood vessels, to go rogue, becoming sticky and adhesive to passing white blood cells.
This kicks off a parade of monocytes migrating into the arterial walls, where they morph into inflammatory macrophages and foam cells, setting up shop inside the vessel wall itself [38, 39]. Meanwhile, smooth muscle cells respond by proliferating and secreting matrix-degrading enzymes that destabilize the plaques. It’s a recipe for progressive arterial damage. Clinical studies have linked higher CRP and IL-6 levels in older adults to increased risks of heart attack and stroke [40].
Chronic inflammation doesn’t stop at the vessel walls. In the heart itself, it drives fibrosis, stiffening cardiac tissue and weakening the heart’s ability to pump blood [41]. It also promotes oxidative stress, triggering endothelial cell death and cutting down nitric oxide availability, which impairs blood vessels’ ability to relax and dilate [42]. Experiments in old rats have shown that blocking TNF-α with drugs like etanercept can improve blood vessel function and reduce oxidative damage [43]. Left unchecked over decades, even mild inflammation cranks up blood pressure, wears out the heart, and sets the stage for clotting events.
Metabolic tissues, namely the muscle, fat, and liver, don’t fare much better. Inflammaging acts like a catabolic force, chewing through muscle mass and metabolic resilience. Sarcopenia, the gradual decline in muscle mass and strength with age, is fueled in part by elevated TNF-α and IL-6, which can directly drive muscle protein breakdown and hobble muscle regeneration [44, 45]. These cytokines activate inflammatory pathways like NF-κB inside muscle fibers, sabotaging insulin signaling and anabolic processes, leading to atrophy and insulin resistance. Unsurprisingly, higher IL-6 levels are strongly linked to frailty and weaker muscle function among the elderly [45].
Fat tissue becomes inflamed with age as well, especially visceral fat. Aging adipose tissue secretes more inflammatory molecules known as adipokines, and macrophages accumulate inside fat depots, releasing TNF-α and IL-1β into circulation. The result is a system-wide spread of insulin resistance, a key step toward the development of Type 2 diabetes [46, 47].
The liver, sitting at the crossroads of metabolism and immune surveillance, doesn’t escape the fallout either. Chronic inflammation promotes fat accumulation and fibrosis in the liver, driving non-alcoholic fatty liver disease (NAFLD), now increasingly common in aging adults [48]. Even the delicate quality control machinery inside cells starts to erode: TNF-α and interferon-gamma can alter the function of proteasomes, shifting them into an inflammatory mode and impairing the normal breakdown of damaged proteins. Over time, misfolded proteins pile up, tipping cells closer to dysfunction [49].
Across these tissues, inflammaging quietly rearranges the cellular landscape, sapping strength, stiffening organs, and muddying the delicate balance that keeps the body resilient.
Effects on the brain, muscles, and other organs
The brain, cushioned behind the blood-brain barrier, is supposed to be a sanctuary from the rough-and-tumble world of systemic inflammation. However, inflammaging has a way of slipping through even the tightest defenses. Chronic systemic inflammation affects the brain both indirectly by weakening the blood-brain barrier and directly through the brain’s own immune sentinels, the microglia. Neuroinflammation becomes a prominent feature of aging, with high levels of peripheral cytokines like IL-6 and TNF-α linked to an increased risk of cognitive decline and dementia across multiple longitudinal studies [50].
These cytokines can disrupt the blood-brain barrier itself, making it more permeable and allowing harmful molecules to seep into brain tissue [51]. Meanwhile, the microglia, which are normally a highly disciplined immune force, become “primed” with age, releasing an excess of inflammatory mediators that can injure neurons or interfere with their function. Chronic microglial activation has been heavily implicated in Alzheimer’s disease, where inflammatory microglia can worsen the accumulation of amyloid plaques and tau tangles. Imaging studies and autopsies of cognitively impaired elderly brains consistently show higher levels of inflammatory markers [52].
Hints from epidemiology once suggested that long-term use of anti-inflammatory drugs like NSAIDs might lower Alzheimer’s risk, though clinical trials have produced mixed results [53]. Still, the basic idea remains tantalizing, and new anti-inflammatory strategies, including NLRP3 inhibitors and immune modulators, are now being explored as adjunct therapies for neurodegenerative diseases. Beyond dementia, chronic inflammation may also play a role in other neural problems that come with aging, including depression and slowed nerve regeneration, by subtly rewiring neurotransmitter systems and damaging neural networks [50].
The musculoskeletal system doesn’t escape such flames either. Inflammation gnaws away not just at muscle mass but at the very scaffolding of the body. The thinning and weakening of bones (osteoporosis) is accelerated by inflammatory cytokines like IL-1, IL-6, and TNF-α, which push the balance toward bone resorption by stimulating osteoclasts while inhibiting bone-forming osteoblasts [54]. In postmenopausal women, the rise in inflammation adds fuel to already declining bone density [55].
Osteoarthritis, long considered a simple mechanical “wear and tear” issue, has increasingly revealed its inflammatory underbelly. Mechanical stresses may initiate cartilage breakdown, but it’s cytokines like IL-1β and TNF-α, produced by senescent chondrocytes and infiltrating macrophages, that sustain the joint damage and keep pain and swelling alive [55]. Over time, chronic inflammation strips away cartilage, stiffens joints, and reduces mobility, feeding into the cycle of frailty that often defines advanced age.
The kidneys, lungs, and even skin are not spared. In the kidneys, persistent inflammation drives fibrosis and chips away at filtration ability, helping explain the steady decline in kidney function seen with age. In the lungs, inflammaging can worsen diseases like chronic obstructive pulmonary disease (COPD) or simply reduce resilience against respiratory insults [56]. Even the skin, the body’s outermost barrier, feels the slow burn of aging inflammation. Chronic low-grade inflammation impairs skin repair, thins the dermal layers, and contributes to the textbook hallmarks of aging skin: dryness, fragility, and delayed wound healing [57].
No organ system gets a free pass. The slow but relentless march of inflammaging weaves itself into every tissue, quietly degrading function long before overt disease becomes visible.
Across all these systems, one common thread weaves its way through the chaos: a slow, persistent inflammatory environment that gradually erodes tissue structure and function. It’s the difference between a fire that scorches a house down in one night versus a smoldering ember that, over years, quietly eats away at the foundation. Chronic, low-grade inflammation doesn’t usually announce itself with fireworks; it wears tissues down molecule by molecule. Many of the chronic diseases we associate with aging are, in large part, the final score of this long, silent battle. Scientists now strongly suspect that inflammaging isn’t just another side effect of aging; it’s a major driver of multimorbidity, the grim parade of overlapping chronic diseases and frailty that shadows old age.
Inflammation and age-related diseases
Chronic inflammation acts like a dark river running underneath the diverse symptoms of aging. Epidemiological studies have repeatedly shown that people with greater levels of inflammatory biomarkers during mid-life or later life face a much greater risk of major age-related diseases, including cardiovascular disease, dementia, cancer, and diabetes [58].
Heart disease and stroke remain two of the top killers in older populations, and chronic inflammation now sits alongside cholesterol and high blood pressure as a major culprit. As discussed earlier, inflammatory processes aren’t just bystanders in atherosclerosis; they actively drive it. Prospective studies consistently show that people with elevated CRP or IL-6 levels have a higher risk of heart attacks and strokes, even after controlling for traditional risk factors like smoking or hypertension [59].
Inflammation plays every position in the atherosclerosis game. It kicks off the initial damage by undermining endothelial function, it encourages plaques to grow, and it throws the final punch when unstable plaques rupture and clot, causing the heart attacks and strokes that land people in emergency rooms. Inflammation also quietly reshapes the heart muscle itself, promoting fibrosis and hypertrophy, setting the stage for heart failure over time.
One landmark piece of evidence tying inflammation directly to heart disease came from the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). For decades, researchers had suspected that inflammation was a co-conspirator rather than an innocent bystander in cardiovascular disease, and CANTOS put that theory to the test. Over 10,000 patients who had already survived heart attacks but still had elevated inflammation levels were recruited. Instead of targeting cholesterol, CANTOS targeted inflammation head-on using a monoclonal antibody against IL-1β called canakinumab [60, 61].
The results were groundbreaking. Patients who received canakinumab had significantly fewer heart attacks and strokes compared to those on placebo, especially among those who achieved the biggest drops in IL-6 levels. In plain terms, reducing inflammation instead of cholesterol or blood pressure lowered the risk of major cardiovascular events. This trial, published in 2017, delivered the first significant evidence in humans that inflammation isn’t just collateral damage in heart disease; it’s an active driver [60, 61].
As a bonus twist, the CANTOS trial stumbled across another tantalizing hint: patients receiving canakinumab also had a reduced incidence of lung cancer. Reducing chronic inflammation, it turns out, might offer broader protective effects than anyone initially guessed [62].
Beyond heart attacks and strokes, chronic inflammation quietly sabotages the heart in other ways too. Atrial fibrillation, the most common arrhythmia in older adults, has been linked to elevated inflammation markers like CRP. Heart failure with preserved ejection fraction (HFpEF), a frustratingly common type of heart failure in the elderly, appears driven at least partly by systemic inflammation stiffening the heart muscle and blood vessels [63].
Although canakinumab isn’t widely used in practice due to high costs and concerns about infection, the success of CANTOS lit a fire under the search for safer anti-inflammatory strategies. Trials are now exploring whether cheaper, existing drugs like low-dose colchicine or even methotrexate might offer cardiovascular protection by dousing vascular inflammation [64, 65]. The message from cardiovascular disease is clear. Years of moderate, untreated vascular inflammation don’t just inflame arteries; they slowly, methodically destroy the heart.
Beyond Alzheimer’s disease, other neurodegenerative conditions show the fingerprints of chronic inflammation too. In Parkinson’s disease, overzealous microglia and systemic inflammation have been linked to faster progression of motor symptoms [66, 67]. Peripheral inflammation doesn’t just haunt the brain’s elite functions; it’s tied to slower walking speed, higher rates of depression [50], and other geriatric syndromes that chip away at autonomy in old age. Put simply, chronic inflammation ages the brain itself. Neuroinflammaging has thus become one of the most dynamic frontiers of research, with the hope that taming brain inflammation early might one day stall or even prevent neurodegeneration.
Cancer
If aging is the number one risk factor for cancer, chronic inflammation is its constant backstage hand. It’s been known for a long time that certain chronic inflammatory conditions pave the road toward malignancy. Ulcerative colitis boosts colon cancer risk [68], hepatitis C leads to liver cancer [69], and chronic gastritis escalates into stomach cancer. The concept of smoldering inflammation as an incubator for tumors has even been enshrined as one of the official hallmarks of cancer itself, thanks to Hanahan and Weinberg [70].
In the context of aging, even low-level systemic inflammation seems enough to lay the groundwork. Epidemiological studies consistently find that individuals with higher CRP or IL-6 levels are more likely to develop cancer and tend to have worse outcomes once cancer strikes [71]. Globally, about 20-25% of all cancers are estimated to have roots in chronic infection or inflammation [72], and when account for the “sterile” inflammation of old age, that number may be even higher.
Chronic inflammation rolls out the red carpet for cancer through several overlapping mechanisms. First, it damages DNA. Inflammatory cells generate reactive oxygen and nitrogen species that bash neighboring cells’ DNA into submission, causing mutations [73]. Over years, this builds up a cellular rogue’s gallery primed for malignant transformation.
Inflammation also impairs the DNA repair systems themselves; chronic exposure to TNF-α or IL-1β can hobble mismatch repair and nucleotide excision repair, allowing errors to slip through [74]. Worse yet, inflammatory signaling can induce rogue enzymes like activation-induced cytidine deaminase (AID) in non-immune cells, leading to targeted, high-efficiency mutagenesis, which is a particular problem in inflammation-associated cancers like colitis-driven colorectal cancer [75].
Second, inflammation showers cells with growth signals. Cytokines like IL-6, IL-1, and TNF-α activate pro-survival pathways like NF-κB and STAT3, promoting proliferation and resistance to apoptosis. IL-6, for instance, can hijack the STAT3 pathway in epithelial cells, turning them into hyperactive replicators [76]. TNF-α doesn’t just inflame, it acts as a tumor promoter, aiding angiogenesis and encouraging mutant cells to migrate [77]. Chronically inflamed tissues endure cycles of cell death and frantic, compensatory proliferation: a perfect storm for expanding mutated clones into full-blown tumors.
Chronic inflammation doesn’t just help initiate cancer; it shapes the battlefield in ways that favor the enemy. In theory, the immune system should detect and destroy emerging tumor cells before they can cause trouble. In practice, chronic inflammation bends the rules.
Over time, the immune environment inside inflamed tissues skews toward a protumor configuration. Instead of sharp-eyed cytotoxic T cells patrolling the area, you get a surge of M2-polarized macrophages: cells that secrete growth factors, remodel the extracellular matrix, and generally act like bad landlords, encouraging tumor growth. T regulatory cells, which normally keep the immune system from overreacting, proliferate and start suppressing the very T cells that could have killed cancerous cells in the first place. Cytokines like TGF-β and IL-10, abundant in chronically inflamed environments, throw another wet blanket on anti-tumor immunity. What you end up with is an immunosuppressive niche that actually helps early-stage tumors hide, thrive, and eventually dominate [78].
Tissue remodeling under chronic inflammation also plays a role. Enzymes like matrix metalloproteinases (MMPs) chew through the extracellular matrix and basement membranes, the structural barriers that normally keep cells confined to their proper neighborhoods. Once those barriers are weakened, cancer cells have a freeway for invasion and metastasis [79]. Chronic inflammation also stimulates the growth of new blood vessels, which are intended to bring in more immune cells for healing but are readily hijacked by tumors to feed their unchecked expansion [80].
In older adults, the fingerprints of inflammaging can be seen on the pattern of cancer emergence. Lung cancer risk skyrockets in people with a history of chronic lung inflammation, whether from smoking, COPD, or even low-grade environmental exposures. Colon cancer in aging populations may partly stem from decades of inflammatory diets, shifts in the gut microbiome, and silent, ongoing colitis. Even cancers that weren’t traditionally linked to inflammation, like certain leukemias, are now tied to chronic low-grade inflammation in the bone marrow. This environment fosters clonal hematopoiesis, the expansion of pre-leukemic mutant clones that sometimes tip into full-blown blood cancer [81].
Worse still, chronic inflammation acts as a brutal evolutionary filter. It selectively pressures cells toward traits that favor survival under stress, which include the loss of p53 tumor suppression. In the liver, in the skin, and in the colon, repeated cycles of inflammatory injury don’t just wear tissue down, they sculpt it into a breeding ground for malignant, resilient cells [82].
Another fascinating clue about the power of inflammation came straight out of the CANTOS trial. As mentioned earlier, patients who received the IL-1β inhibitor not only suffered fewer cardiovascular events, they also showed a striking reduction in lung cancer incidence and mortality compared to the placebo group. This wasn’t something that researchers had set out to prove; it emerged as an unexpected bonus. These findings have since sparked new clinical trials to see whether blocking inflammation could deliberately prevent or slow down certain cancers [83, 84].
Elsewhere, similar hints emerge. In patients with type 2 diabetes, which is a metabolic condition marinated in low-grade inflammation, the use of anti-inflammatory agents like aspirin has been associated with a reduced risk of cancer in some studies, further supporting the idea that cutting down chronic inflammation might also starve one of cancer’s crucial fuels [85].
Overall, chronic inflammation creates a grimly fertile environment for cancer to take root and thrive. It’s as if inflammaging doesn’t just lay down the kindling for malignancy; sometimes, it strikes the match as well. That’s why anti-inflammatory strategies are now being explored not just as treatments but as cancer prevention tools, especially in older populations. Others are looking at IL-6 or TNF-α blockers as potential therapies to slow cancer progression or treat cancer-related wasting syndromes like cachexia [13]. Inflammation is not the sole architect of cancer, but it’s a key subcontractor that speeds up the project.
Therapeutic interventions
Given how thoroughly chronic inflammation is entangled with aging, it’s no surprise that a wide array of therapeutic strategies are being pursued to cool the fires of inflammaging. Some approaches borrow existing tools originally developed for autoimmune diseases; others push into experimental territory, while still others circle back to deceptively simple lifestyle interventions. The common goal: dampen the slow burn of chronic inflammation, improve tissue health, and extend the number of years we spend functional rather than fragile.
Anti-Inflammatory medications
Targeting inflammatory cytokines has already proven a game-changer for chronic inflammatory diseases, and there’s growing hope that these strategies could be adapted for aging itself. One of the clearest examples is the class of TNF-α inhibitors. TNF-α, a heavyweight pro-inflammatory cytokine, reliably rises with age, and biological drugs like etanercept, infliximab, and adalimumab have been used for decades to neutralize it in diseases like rheumatoid arthritis, psoriasis, and inflammatory bowel disease.
These drugs taught an important lesson: block a single inflammatory signal, and you can achieve widespread systemic benefits. In rheumatoid arthritis, TNF-α inhibitors don’t just cool down the joints; they lower CRP levels, trim systemic inflammation, and, intriguingly, seem to reduce the risk of heart attacks compared to patients left smoldering with persistent inflammation. In aged animal models, knocking down TNF-α signaling improved vascular function, producing significant rejuvenation effects in blood vessels [86, 87].
Naturally, the fantasy is that one day, low-dose or intermittent cytokine inhibition could be tested in older adults not to treat existing disease, but to pre-empt it, tamping down baseline inflammation before it chisels away at tissues. However, there’s a catch: TNF-α and its cytokine cousins aren’t just pyromaniacs; they’re part of the fire department too. Blocking them raises infection risks, a particular concern for elderly people who are already riding the thin line of immune competence. Any broad use would require a careful weighing of benefits against the risk of tipping the immune system too far into helplessness [88].
Beyond TNF-α, the cytokine inhibition menu includes IL-1 and IL-6 antagonists. Anakinra, an IL-1 receptor blocker, has found a home in rare autoinflammatory diseases and small frailty trials in older adults, where it showed modest improvements in inflammation markers and anemia [89]. Canakinumab, the star of the CANTOS trial, didn’t just lower heart attacks, it also slashed lung cancer incidence [83], cementing its reputation as a potential geroprotector, even if it’s currently trapped behind a paywall of astronomical cost. Tocilizumab, an IL-6 receptor blocker, is another contender, used in rheumatoid arthritis [90].
All this suggests that neutralizing key inflammatory cytokines can, at least temporarily, break the cycle of inflammaging. But none of these drugs are ready for prime-time anti-aging prescriptions yet. They’re expensive, and they blunt the immune system in ways that could backfire if used indiscriminately. Ongoing research is trying to pinpoint subsets of older adults, such as those with sky-high IL-6 levels and looming cardiovascular risks, who might benefit most from targeted interventions.
Meanwhile, the old warhorses of inflammation control, such as NSAIDs and corticosteroids, remain less-than-ideal solutions. NSAIDs can turn down inflammatory pathways, but chronic use shreds the gut lining, stresses the kidneys, and paradoxically increases cardiovascular risk [91]. Corticosteroids, while nuclear options against inflammation, come with their own monstrous baggage, including but not limited to bone loss, diabetes, immune suppression, and cognitive side effects [92].
Low-dose aspirin straddles a gray zone. Its anti-inflammatory effects, coupled with its blood-thinning properties, have made it a sometimes-hero in reducing second heart attacks and, perhaps, in nudging down colon cancer risk. However, recent large trials in healthy older adults have been sobering, with no clear mortality benefit and a higher risk of major bleeding events. Aspirin, it turns out, isn’t the free ride some hoped it would be [93].
What all these experiences highlight is simple: the tools we have now are either too blunt or too risky for mass deployment against inflammaging. The future will demand safer, smarter anti-inflammatories: precision strikes instead of carpet bombing.
One promising direction for dialing down inflammation without throwing the immune system into chaos is to use drugs that modulate inflammatory signaling more subtly, rather than going in with a biochemical sledgehammer.
Statins, for instance, are best known as cholesterol-lowering agents, but it turns out they also have modest anti-inflammatory effects, such as lowering levels of inflammatory cytokines and CRP. Some researchers think that this “side talent” partly explains why statins do such a good job at reducing cardiovascular events, even beyond their effects on LDL cholesterol [94]. Then there’s low-dose methotrexate, a drug used to suppress the immune system in rheumatoid arthritis. Hopes were high that it might also prevent heart attacks by reducing inflammation. However, when put to the test in the CIRT trial, it flopped, with no meaningful drop in IL-6 or CRP and no cardiovascular benefit. Biology, as usual, remains stubbornly more complicated than we’d like [95].
Another intriguing candidate is the family of JAK inhibitors, like baricitinib and ruxolitinib. These drugs block Janus kinase enzymes, key players in the transmission of inflammatory signals. JAK inhibitors are already used in diseases like rheumatoid arthritis and certain blood cancers, and because they target multiple cytokine pathways at once, there’s it’s an appealing way of tamping down broad inflammatory tone without having to block each cytokine individually. Early clinical studies have found that JAK inhibitors can indeed reduce inflammatory biomarkers. Again, the usual devil lurks; suppressing the immune system too much causes infection risks to climb [96]. Overall, the use of existing anti-inflammatory drugs offers proof that dialing down inflammation can impact disease risk.
Experimental approaches
Because the NLRP3 inflammasome sits at the molten core of inflammaging, a lot of excitement now surrounds the idea of blocking it directly. One prototype compound is MCC950, also known as CRID3, a small molecule that clamps down on NLRP3 activation before it can kick off the inflammatory cascade. In preclinical studies, MCC950 has shown impressive ability to lower IL-1β and IL-18 levels and ease conditions like arthritis, metabolic syndrome, and even neurodegenerative pathologies [97].
When given to middle-aged mice, NLRP3 inhibitors didn’t just tamp down inflammation, they preserved cognitive function well into old age, suggesting a broader systemic benefit beyond any single organ. Dapansutrile (OLT1177) is another NLRP3 inhibitor making its way through clinical trials, initially tested in gout and heart failure. Administered orally, it’s shown promising early results in reducing IL-1β and pain in gout patients [98, 99].
The hope is that these drugs might eventually find a home in broader geroprotection strategies, essentially turning down inflammaging at its molecular source.
Another way to hit the inflammasome system is to block its outputs. Canakinumab, the IL-1β antibody from the CANTOS trial, isn’t specific to NLRP3 but effectively neutralizes one of its main inflammatory missiles. Researchers are also tinkering with drugs that target IL-18, though its exact role in aging isn’t as well fleshed out as IL-1β. Caspase-1 inhibitors are in development to stop the inflammasome’s key enzyme in its tracks, and some teams are exploring ways to block gasdermin D, the protein that punches holes in cell membranes to execute inflammatory cell death (pyroptosis) [100].
All of these strategies are still experimental, but they reflect a bigger shift in aging research: moving beyond trying to fix the downstream damage of inflammaging and instead cutting off the upstream sparks before the fire takes hold.
Beyond targeting inflammasomes directly, researchers are widening the net. Other inflammatory pathways have emerged as potential targets, such as STING, the cytosolic DNA-sensing pathway that ramps up inflammation in response to misplaced genomic fragments, a common occurrence as genomic instability worsens with age [101]. Toll-like receptors (TLRs), ancient microbial detectors, also become hypersensitive in aging tissues, contributing to the chronic drumbeat of inflammation [102]. Even metabolic regulators like AMPK (adenosine monophosphate-activated protein kinase), usually celebrated for their role in energy balance, are being eyed for their ability to put a lid on inflammatory signaling [103].
Some researchers are flipping the script entirely, asking not how to suppress pro-inflammatory forces but how to amplify the body’s natural anti-inflammatory defenses. Boosting IL-10, an anti-inflammatory cytokine, or expanding populations of regulatory T cells could theoretically tilt the balance back toward equilibrium. In mouse models, systemic overexpression of IL-10 has reduced age-related inflammation and frailty, suggesting that sometimes the answer isn’t just silencing bad signals but nurturing the good ones [104].
One of the most intriguing experimental strategies is the use of senolytics, drugs that selectively clear out senescent cells, the professional rabble-rousers of the inflammatory microenvironment. Agents like the dasatinib and quercetin combination [105, 106] and the naturally occurring flavonoid fisetin have shown remarkable effects in animal studies. Administering senolytics to aged mice doesn’t just lower tissue levels of SASP factors; it rejuvenates blood vessel elasticity, improves muscle regeneration, and enhances overall function.
Early-phase human trials in conditions like idiopathic pulmonary fibrosis and diabetic kidney disease have started to show reductions in systemic inflammation after senolytic treatment. If these therapies prove safe and scalable, we may one day see periodic senolytic “flushes” prescribed to sweep out pockets of cellular rot before they cause broader organ decline [31].
Another promising angle is tackling the gut microbiome. Aging tends to scramble the microbial lineup in our intestines, favoring species that increase gut permeability and leak inflammatory molecules like lipopolysaccharide (LPS) into the bloodstream. Inflammatory seepage from the gut then fans out systemically. Rodent studies have shown that restoring a youthful gut microbiota, such as via probiotics, prebiotics, or even fecal transplants from young mice, can lower inflammatory cytokine levels and improve metabolic health [107]. In humans, strategies like enhancing short-chain fatty acid production through diet or supplements could be a low-risk way to dial down systemic inflammation without heavy pharmaceutical artillery [108].
There’s also a metabolic side of the equation. NAD⁺ boosters like nicotinamide riboside (NR) and NMN have captured attention for their ability to recharge declining NAD⁺ pools in aging tissues. Since sirtuins like Sirt2 and Sirt3, enzymes that put brakes on inflammation and oxidative stress, depend on NAD⁺, replenishing it could act as a systemic anti-inflammatory tune-up [109]. In aging animal models, raising NAD⁺ levels not only rejuvenates mitochondrial function but also dials down tissue inflammation [110]. Similarly, metformin, a venerable diabetes drug and AMPK activator, is being tested in the Targeting Aging with Metformin (TAME) trial, partly based on its promise to lower chronic inflammation and delay the onset of multiple age-related diseases [111].
It’s not hard to imagine a future in which checking inflammatory biomarkers in older adults becomes as routine as monitoring cholesterol or blood pressure today. In the future, physicians might prescribe a cocktail of interventions, such as clearing senescent cells, tweaking the microbiome, and boosting NAD⁺, to hold back the slow burn of inflammaging. If the research pans out, this could be the beginning of a new era: not just extending lifespan, but preserving the quality of those extra years.
Lifestyle and behavioral interventions
While high-tech therapies and molecular sniper shots are thrilling to imagine, some of the most powerful weapons we already have against inflammaging require nothing more exotic than sneakers, persistence, and maybe a little willpower. Lifestyle modifications remain a cornerstone because they don’t just target one pathway or molecule; they hit multiple levers of aging biology at once, often with bonus side effects like better mood, stronger muscles, and a lower risk of dying prematurely.
First and foremost is exercise. Regular physical activity is arguably the single most potent, consistently proven anti-inflammatory intervention known to modern medicine. Study after study shows that both aerobic exercise, such as walking, jogging, and cycling, and resistance training, including good old-fashioned weightlifting, can lower systemic inflammation in older adults [112, 113].
In one randomized trial, elderly participants who committed to 12 months of moderate-intensity exercise showed significantly lower blood IL-6 levels compared to their sedentary peers [114]. This wasn’t a small blip. Exercise shifted their entire inflammatory profile closer to that of a much younger population [114].
Exercise melts away visceral fat, the deep abdominal fat depot that acts like a malevolent endocrine organ, spitting out inflammatory adipokines. It stimulates muscle cells to release anti-inflammatory molecules called myokines, such as IL-10 and IL-1 receptor antagonist (IL-1ra), which act like tiny peace treaties in the bloodstream. It even rewires the immune system itself, increasing the number of regulatory immune cells that help tamp down inflammation [115, 116].
Perhaps the most stunning evidence comes from studies of lifelong exercisers. In one investigation, older adults who had remained highly active throughout their lives had IL-6 levels that barely budged compared to their sedentary counterparts, almost as if exercise had partially “immunized” them against the slow, relentless rise of inflammation that aging usually brings [117].
Even short-term interventions in previously sedentary older adults have been shown to reprogram inflammatory gene expression, improve endothelial function, and nudge the immune system toward a calmer, better-behaved state [118].
The anti-inflammatory powers of exercise are one major reason why physical activity lowers the risk of heart disease, diabetes, and cognitive decline. These benefits aren’t disconnected; they converge through a common pathway: reducing chronic inflammation [117].
The current public health gospel is clear: at least 150 minutes of moderate-intensity exercise per week for older adults. Enough to sweat, enough to breathe a little harder, enough to tip the molecular scale back toward resilience instead of degeneration [119].
Diet
If exercise is the low-hanging fruit of anti-inflammaging, diet is the soil that nourishes or poisons the whole tree. Food exerts a profound influence on inflammatory tone, shaping the gut microbiota, oxidative stress levels, and systemic immune signaling.
On one end of the spectrum, diets rich in refined sugars, processed fats, and ultra-processed foods act like fertilizer for inflammation. They drive oxidative stress, disrupt the gut microbiome, and activate inflammatory pathways like NF-κB with a vengeance [120, 121]. On the other end, diets built on healthier foods can do the opposite, acting like molecular firefighters [120].
The Mediterranean diet, for example, is famous for its liberal use of olive oil, fish, nuts, and leafy greens. In observational studies, older adults who stuck closer to a Mediterranean-style eating pattern had significantly lower CRP levels compared to people chowing down on a typical Western diet. It’s not just one magic ingredient. The Mediterranean diet is a dense package of anti-inflammatory artillery: omega-3 fatty acids that give rise to inflammation-resolving molecules like resolvins and protectins, polyphenols and antioxidants that cool oxidative stress and downregulate NF-κB activation, and fiber that feeds a healthier, less inflammatory gut microbiome [122, 123].
Clinical trials have confirmed that switching to a Mediterranean diet, or even supplementing key components like extra-virgin olive oil, can reduce inflammatory cytokines and improve endothelial function [124].
Weight loss through calorie control can further dial down inflammation, especially when it peels away visceral fat. Studies show that losing just 5-10% of body weight in overweight older adults is enough to lower IL-6 and CRP significantly. Inflammation, it turns out, is tightly tied to adiposity, particularly the kind that wraps itself around internal organs like a biological boa constrictor [125].
Fiber boosts short-chain fatty acid production by gut microbes. Undoing deficiencies in vitamins C, D, and E restores immune balance. Curcumin, the active compound in turmeric, has notable NF-κB-suppressing abilities. Though supplements like curcumin and resveratrol are still under investigation, they offer tantalizing hints at future dietary fine-tuning for inflammation control [126].
Importantly, avoiding chronic overnutrition matters. Persistent nutrient overload overstimulates pathways like mTOR, which in turn provoke innate immune cells into a state of low-grade inflammatory buzz [127]. Eating the right amount of food makes the immune system less likely to misfire.
Caloric restriction and intermittent fasting
Perhaps no intervention has captured the imagination of aging researchers more than caloric restriction (CR). In animal models, cutting calories by 20-30% while avoiding malnutrition lifespan to stretch out like a road with fewer potholes [128].
CR might be accomplishing this through slashing chronic inflammation. Calorically restricted rodents and monkeys show lower levels of systemic cytokines, less tissue NF-κB activation, and an overall anti-inflammatory gene expression profile [129].
In humans, the CALERIE trial, an ambitious, controlled foray into long-term caloric restriction, found that cutting calories led to measurable declines in TNF-α, CRP, and metabolic inflammation markers [130]. Even short-term experiments with intermittent fasting or time-restricted eating show promising anti-inflammatory effects, likely through improvements in metabolic resilience and cellular stress responses that tip the immune system toward a quieter baseline [131].
Mechanistically, caloric restriction seems to activate resilience pathways like AMPK and sirtuins, molecular guardians that also keep inflammation in check [132].
However, full-blown caloric restriction might not be practical or safe for all older people. Nutritional needs, muscle preservation, and frailty risk all complicate the picture. Mild, strategic versions, such as periodic fasting-mimicking diets, could offer a way to periodically “reboot” inflammation without risking malnutrition [133].
In sum, how much and what we eat shapes immune tone, gut composition, and oxidative balance. Ultimately, it controls how fast or slow the inflammatory clock ticks inside our tissues.
Stress reduction and sleep
Inflammaging cannot be understood without including the quiet saboteurs of stress and bad sleep. Chronic psychological stress, whether from caregiving, financial worries, existential dread, or just the endless grind of modern life, raises systemic inflammatory markers such as IL-6. It’s like living under a slow intravenous drip of cortisol and inflammatory cytokines.
The good news is that mind-body practices like meditation, tai chi, and yoga aren’t just feel-good hobbies; they leave molecular fingerprints. In elderly participants, such mindfulness interventions have been shown to lower CRP levels and even shift inflammatory gene expression profiles toward a calmer state [134].
Poor sleep quality or sleep disorders like apnea crank up inflammatory signaling. Even a few nights of fragmented sleep can elevate cytokines like IL-6 and TNF-α. Fixing sleep hygiene or treating apnea doesn’t just make you feel better, it literally dials down systemic inflammation at the source [135].
Smoking and alcohol
Of course, no lifestyle discussion would be complete without obligatory but vital reminders. Smoking is a full-blown inflammatory disaster. It doesn’t just wreck lungs; it primes the vascular endothelium for atherosclerosis, jacks up CRP and IL-6, and leaves a smoldering signature of inflammation throughout the body. Quit smoking, and over time, these inflammatory markers fall [136].
Alcohol is a trickier story. In modest doses, such as a glass of red wine tucked into the Mediterranean diet, it might exert mild anti-inflammatory effects. However, heavy drinking flips the script entirely, fueling inflammation, gut permeability, and immune suppression. The rule of thumb: moderation if at all, and never with the idea that more is better [137].
Lifestyle synergy
Taken together, healthy lifestyle choices create an internal ecosystem that’s less prone to chronic inflammation. Exercise acts like a slow-release anti-inflammatory drug. Diet serves as a daily regulator of oxidative stress and gut integrity. Stress management and good sleep insulate the nervous and immune systems from hyperactivation. Quitting smoking and moderating alcohol aren’t just about living longer, they’re about maintaining molecular peace inside aging tissues.
Better yet, these interventions synergize. Combining exercise and healthy eating doesn’t just add benefits, it multiplies them, creating a compounding reduction in CRP and other inflammatory markers [138]. Unlike pharmaceuticals, which often target single molecules, lifestyle interventions address the complex, tangled web of upstream causes: visceral fat, microbial imbalances, cortisol overload, nutrient stress.
Until the arrival of bespoke anti-inflammaging drugs, lifestyle optimization remains the front line. Arguably, even when those drugs come, lifestyle will still be the ground on which healthy aging is either built or quietly undermined.
Involvement with other hallmarks of aging
Of course, chronic inflammation doesn’t exist in some molecular vacuum. It’s enmeshed in a dense web of cause-and-effect with the other hallmarks of aging. Like a bad actor in an ensemble cast, it both triggers other forms of biological decline and is fed by them in return [139].
Genomic instability
There is an unholy marriage between inflammaging and genomic instability. Chronic inflammation isn’t just an innocent bystander watching DNA fall apart; it helps drive the destruction. Inflammatory cells generate reactive oxygen species (ROS) and reactive nitrogen species (RNS), which bombard nearby DNA with mutagenic insults [140]. Meanwhile, inflammatory cytokines don’t merely yell at cells, they actively sabotage DNA repair crews. Long-term exposure to these signals downregulates DNA mismatch repair proteins and nucleotide excision repair enzymes, leading to error-prone replication and a buildup of mutations [74].
Inflammation can also activate rogue editing machinery. Enzymes like activation-induced cytidine deaminase (AID), usually tasked with refining immune responses, get mistakenly flipped on in somatic cells under chronic inflammatory stress, creating a mutagenic mess [141].
However, this is not a one-way street. Genomic instability can stir the inflammatory pot right back. DNA damage triggers cellular stress responses that converge on NF-κB, the same master switch that drives pro-inflammatory gene expression [142]. Cells unable to patch up their genomes may drift into senescence and secrete inflammatory factors as part of the SASP. Even worse, if bits of nuclear DNA or retrotransposon-derived sequences leak into the cytoplasm, which happens more often as genomic maintenance falters, they activate the cGAS-STING pathway, unleashing interferons and inflammatory cascades [101].
It’s a feedback loop worthy of Greek tragedy: damage begets inflammation, inflammation deepens the damage, and together, they spiral cells ever closer to dysfunction, cancer, and aging.
Telomere attrition
Telomeres are the protective caps on chromosomes that get shorter with every cell division, ticking down the biological clock. It turns out that inflammation has no qualms about taking a chainsaw to the hourglass. Inflammatory cytokines like TNF-α and IL-6 have been associated with shorter telomeres in humans. Mechanistically, TNF-α can inhibit telomerase, the enzyme responsible for topping off telomere length, accelerating the erosion of chromosomal ends [143]. Oxidative stress, a byproduct of chronic inflammation, singles out telomeres for disproportionate damage; and their structure makes them particularly sensitive to ROS [144, 145].
The damage isn’t just cosmetic. Critically shortened telomeres activate DNA damage responses that push cells into senescence [146], where they crank up SASP factors, pouring more inflammatory signals into the tissue environment. Mice engineered with short telomeres show elevated systemic IL-6 levels, turning their bloodstream into a low-grade inflammatory stew [147].
Once again, the feedback loop emerges: inflammation hastens telomere attrition, and telomere dysfunction turbocharges inflammation. Left unchecked, this vicious cycle grinds tissues down toward frailty and disease.
Epigenetic alterations
Inflammatory cytokines, for example, can distort DNA methylation landscapes. Long-term exposure to IL-6 or CRP has been linked to hypomethylation at repetitive DNA elements, while inflammation-induced nitric oxide synthase (iNOS) can inhibit DNA methyltransferase 1 (DNMT1), promoting demethylation of retrotransposons, which are ancient genomic hitchhikers that are better left silenced [148, 149].
Histone modifications don’t escape either. NF-κB, ever the mischief-maker, recruits histone acetyltransferases to inflammatory gene promoters, loosening chromatin structure and locking in pro-inflammatory gene expression [150].
Over time, this creates a kind of inflammatory memory at the epigenetic level. Cells remember past inflammatory insults, making them quicker to re-ignite the flames even when the original threat is long gone. Meanwhile, normal age-related epigenetic drift, such as promoter hypomethylation, exposes previously silent transposable elements, activating innate immune sensors like cGAS-STING and triggering yet more inflammation [151].
Intriguingly, there’s evidence that calming inflammation can slow the ticking of epigenetic aging clocks, hinting that inflammaging and epigenetic drift might be co-conspirators rather than separate villains [152].
Loss of proteostasis
Protein quality control, the behind-the-scenes housekeeping that keeps cells tidy, is another system deeply entangled with inflammation. When proteostasis falters and misfolded or aggregated proteins accumulate, they don’t just sit there like forgotten laundry. They act as DAMPs that rattle immune sensors and ignite inflammation. Amyloid plaques in Alzheimer’s disease are a textbook example, activating inflammasomes in microglia and setting off neuroinflammatory cascades [153].
Conversely, chronic inflammation corrupts the very machinery meant to uphold proteostasis. TNF-α and interferon-gamma can induce the formation of immunoproteasomes, a kind of emergency-mode protein shredder that prioritizes antigen processing over regular quality control [154]. These are useful in fighting pathogens but disastrous for maintaining everyday protein homeostasis.
Inflammation also triggers endoplasmic reticulum (ER) stress, an overload of misfolded proteins in the ER, which activates NF-κB and fans the inflammatory flames even higher [155].
Autophagy is a vital process by which cells clean out damaged proteins and organelles. With age, autophagy declines, and inflammation actively accelerates this decay. TNF-α signaling through mTOR blocks autophagy initiation, while IL-1β disrupts autophagic flux [156].
This results in damaged proteins and malfunctioning mitochondria piling up, leaking more inflammatory signals, and driving a self-perpetuating cycle of dysfunction. Encouragingly, enhancing autophagy, whether through mTOR inhibitors, caloric restriction, or other means, has been shown to quell inflammation in aged tissues, revealing just how tightly proteostasis and inflammaging are intertwined [157].
Mitochondrial dysfunction
Mitochondria are not just innocent bystanders in aging, they are heavily armed saboteurs once things go wrong. Old, battered mitochondria start leaking mitochondrial DNA (mtDNA) into the cytoplasm or extracellular space. To immune sensors like the inflammasome and the cGAS-STING pathway, this free-floating DNA screams “danger,” unleashing bursts of IL-1, interferons, and other inflammatory signals [158]. Mitochondrial production of ROS spikes as well, feeding directly into NF-κB activation and inflammatory gene expression.
Inflammation pays the damage forward. Caspase-1 activation during inflammasome firing can cleave proteins like Parkin, a key player in mitophagy, the targeted disposal of bad mitochondria [159]. Disrupting mitophagy leads to a vicious cycle: defective mitochondria accumulate, produce even more ROS and inflammatory mediators, and spiral cells into deeper dysfunction.
Inflammation even rewires immune cell metabolism. Under chronic inflammatory conditions, immune cells like macrophages shift toward glycolysis, a faster but dirtier metabolic program fueled by mTOR activation, further entrenching inflammatory behavior [160, 161].
Efforts to boost mitochondrial health, whether through exercise, NAD⁺ boosters, or AMPK activation, often dampen inflammation in parallel, underlining the point: mitochondrial failure and inflammaging are two sides of the same degenerative coin.
Cellular senescence
Senescent cells are inflammation factories, but the relationship cuts both ways. Chronic inflammation doesn’t just result from senescence; it creates more senescent cells.
Cells exposed to inflammatory cytokines or bacterial endotoxins can be gradually bullied into a senescent-like state, a phenomenon sometimes called secondary senescence. Studies have shown that treating cells with TNF-α or IL-1β over time causes DNA damage and telomere dysfunction, flipping cells into permanent growth arrest. Neighboring cells aren’t safe either: immune cell oxidative bursts, like neutrophils flinging out reactive oxygen species, can induce senescence in nearby innocent bystanders [162, 163].
This leads to a self-perpetuating nightmare: inflammation seeds more senescent cells, which secrete more SASP factors, which feed more inflammation. Breaking this loop has huge therapeutic potential. Experiments show that clearing senescent cells (senolysis) or suppressing their inflammatory SASP output can dramatically lower chronic inflammation in aged tissues and restore some youthful function. In short, inflammaging and cellular senescence aren’t separate fires; they are a single blaze feeding itself [164].
Stem cell exhaustion
Stem cells, the body’s long-term investment accounts, also get hammered by chronic inflammation. Hematopoietic stem cells (HSCs) in the bone marrow are the originators of all blood and immune cells. With age, these cells get weird: they start preferring to produce myeloid cells over lymphoid cells and lose their ability to regenerate themselves effectively. Chronic inflammation plays a starring role in this corruption [165].
Constant exposure to inflammatory cytokines drags HSCs out of their safe, quiescent resting phase. TNF-α and IL-1β, in particular, prod HSCs into cycling and differentiating into myeloid lineages, gradually draining the stem cell pool [166]. Chronic IL-1 signaling has been shown to skew HSC output and exhaust their self-renewal ability; block IL-1, and you can partially preserve youthful HSC function.
IFN-γ, another cytokine elevated in chronic inflammation and chronic infections, also activates HSCs unnecessarily, contributing to their burnout over time [167].
What you get is a bone marrow that looks exactly like it’s been marinated in low-grade inflammation for decades: too many myeloid progenitors, not enough lymphoid ones, and a blood and immune system increasingly prone to dysfunction [168].
It’s not just the bone marrow. Muscle satellite cells, the stem cells responsible for muscle repair, sit in an old, inflamed tissue environment loaded with TNF and TGF-β. Instead of lying low and regenerating muscle when needed, these cells get pushed into premature differentiation or senescence, hobbling muscle regeneration. Neural stem cells in the aging brain face a similar hostile environment, with IL-6 and other inflammatory signals tamping down their ability to generate new neurons [168, 169].
Thus, inflammation accelerates stem cell exhaustion, while dwindling, exhausted stem cell pools make it harder to maintain tissue homeostasis, fueling inflammaging yet again in another harmful loop.
Promisingly, experiments in mice show that dialing down inflammation can partly rejuvenate aged HSCs, suggesting that breaking the cycle between inflammation and stem cell exhaustion might be one route toward healthier aging [170, 171].
Altered intercellular communication
The hallmark of altered intercellular communication is a catch-all for what happens when cells stop talking to each other properly or, worse, start sending the biological equivalent of drunk texts at 3 AM. Chronic inflammation is, in many ways, the poster child for this phenomenon.
Back in 2013, when López-Otín and colleagues first laid out the hallmarks of aging, chronic inflammation was bundled under this umbrella. Now that inflammaging has been promoted to a standalone hallmark, what remains under altered communication is the rest of the messy signaling landscape, and it’s still intimately tied to inflammation [5].
As we age, endocrine signaling goes through its own midlife crisis. Cortisol levels tend to rise, sex steroid levels drop, and these hormonal shifts warp immune system behavior. High cortisol can blunt adaptive immunity while fueling low-grade inflammation, a biological lose-lose. Meanwhile, inflammatory cytokines don’t stay in their lane. They interfere with insulin signaling, contributing to the nutrient sensing deregulation seen in aging, and mess with leptin signaling, which can distort appetite regulation and metabolic homeostasis [172]. The result is an endocrine-immune traffic jam in which hormones and cytokines cross paths in ways that only amplify the problems of aging.
The human gut microbiome is a sprawling microbial city-state. Aging tends to perturb the microbiome, shifting its composition toward species that produce more pro-inflammatory molecules like lipopolysaccharides [173-175]. These bacterial products slip into the bloodstream thanks to a weakened gut barrier, stoking systemic inflammation. In turn, systemic inflammation feeds back to further disrupt gut integrity and microbiota balance, locking host and microbes into yet another vicious cycle [175].
In short, even though chronic inflammation now has its own throne, altered intercellular communication remains deeply entangled with it. Cells, hormones, neurons, and gut microbes all start misfiring and miscommunicating as we age, creating a biological cacophony where once there was orchestration.
In short, chronic inflammation is like a spider at the center of the aging web, twitching its sticky little legs on every strand. Tugging on genomic instability here, jerking telomeres loose there, leaving epigenetic graffiti behind, clogging proteostasis pipelines, exhausting stem cell reserves. The worst part is that the web of damage tugs right back, fueling inflammation, which fuels more damage, in a cycle that’s as efficient as it is destructive [139].
This interconnectedness is precisely why targeting inflammation has such appeal: cut the right thread, and the whole snarled web might sag a little. Some geroscientists now argue that anti-inflammaging strategies could unclog aging biological machinery, rewiring the feedback loops and restoring a little youthful function to the system. If aging is a symphony of breakdowns, then calming the inflammatory section could bring the whole orchestra back into something resembling tune [176].
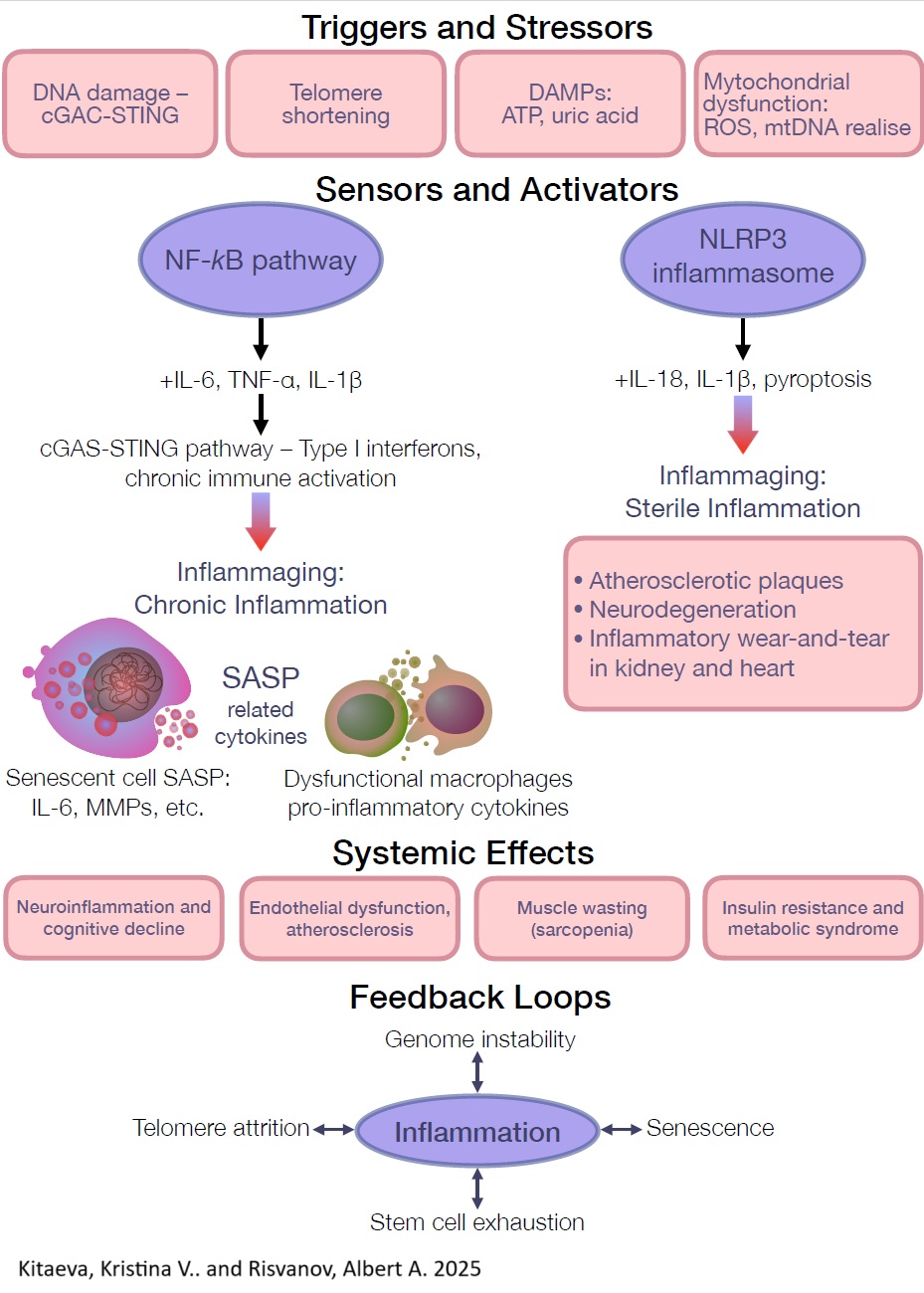
Conclusion and future directions
Chronic inflammation, once just a background hum, has been thrust into the spotlight as a critical, unifying hallmark of aging. Inflammaging isn’t just another nuisance; it’s the slow-burning campfire around which most age-related pathologies huddle. Heart disease, Alzheimer’s, and cancer all find warmth in its glow. Recognizing chronic inflammation as a standalone hallmark [6] is more than a taxonomic upgrade. It’s a redirection of scientific ambition: tame this one process, and it might be possible to tip the aging trajectory itself.
Why is this shift so important? Because it hints at a sort of biological two-for-one deal. Instead of targeting heart disease, Alzheimer’s, and diabetes separately, as if they were independent house fires, we can aim the firehose at the shared arsonist: chronic inflammation. Interventions that lower inflammatory load, whether they be IL-1β blockers, exercise regimens, senolytics, or microbiome tweaks, have shown broad-spectrum benefits: fewer heart attacks, sharper cognition, stronger muscles, and maybe even a little extra time above ground [176]. In the future, anti-inflammaging protocols could become the Swiss Army knives of geriatric medicine: versatile, preventative, and multi-systemic.
Of course, this rosy vision comes with thorns, such as timing. Is it better to strike early, with lifestyle and preventive nudges while the fire is still embers, or later, when the house is already smoldering but still salvageable? Evidence suggests that both approaches may have merit. Preventative strategies like midlife exercise and anti-inflammatory diets could build resistance long before trouble hits, but even late-game interventions, such as canakinumab in seniors with high CRP, can shave off heart attacks [61].
Safety, however, is a bigger, gnarlier thorn. The immune system isn’t just some cantankerous old neighbor causing trouble; it’s also the body’s home security system. Too much suppression, and infections and cancers start taking over. Therefore, the trick is in how to cool smoldering inflammation without dousing the vital flames. Precision interventions, such as targeting pathways like NLRP3 without throwing the whole immune orchestra into chaos, or using intermittent dosing to avoid leaving defenses down for too long, offer the most hope.
Ultimately, understanding inflammaging cracks open the door to interventions that don’t just chase symptoms but actually reset the aging program itself. It’s a thrilling, messy, high-stakes frontier. And if we get it right, the rewards could be enormous: not just a few more years tacked onto life, but a life that feels more like living.
Looking ahead, the future of fighting inflammaging doesn’t look like one magic bullet. It looks like a cocktail: something worthy of a very strange, very expensive happy hour. Imagine a personalized mix of a senolytic agent to flush out inflammatory old cells, an NLRP3 inhibitor to put the innate immune system on a tighter leash, a low-dose cytokine blocker to mop up excess IL-6 or TNF-α, plus a dash of a probiotic or a nutraceutical to nudge the gut and metabolism toward an anti-inflammatory state. Throw in a prescribed exercise and diet plan tailored to molecular quirks, and this might one day be a standard anti-aging regimen. This is ambitious, but trials are already testing combinations, such as combining senolytics with strength training and omega-3 supplementation with aerobic exercise, to see if the anti-inflammatory benefits stack.
Another piece of the future puzzle is biomarkers. In the years ahead, your annual check-up might include an “inflammaging score”: not just CRP and IL-6, but a whole panel of SASP factors, cytokine profiles, and possibly transcriptomic signatures [177]. If your inflammation age (iAge as proposed by Furman and colleagues), is running ahead of your actual years, it could trigger early intervention [178]. This is where preventive medicine gets exciting: rather than waiting for a heart attack or cognitive slide, we might catch inflammaging in mid-flare and squelch it early.
Meanwhile, research is pushing upstream, trying to find what lights the inflammaging fuse in the first place, whether that’s asymptomatic infections to blame, a critical mass of senescent cells, or a slow accumulation of mitochondrial DNA mutations. Understanding the earliest sparks could allow us to prevent the fire before it starts. There’s also rising interest in how aging impairs not just the suppression of inflammation but its resolution. Specialized pro-resolving mediators like lipoxins and resolvins, normally tasked with turning inflammation off, seem to falter with age. Boosting or mimicking these mediators could represent a gentler, more elegant way to end the chronic smolder.
In sum, chronic inflammation isn’t just a symptom of aging; it’s one of the main engines under the hood. However, that gives us leverage. Future strategies will likely mix therapies, such as drugs, biologics, and senolytics, with lifestyle upgrades, such as diet, exercise, and stress reduction, to keep the inflammatory fires of aging at a manageable flicker. What was once treated as a biological afterthought is now seen as one of aging’s prime movers and one of its most actionable vulnerabilities.
With clinical trials underway and new therapies emerging, there’s real hope that inflammaging will go from inevitable to modifiable. Targeting chronic inflammation won’t just help us live longer; it might help us live better, with fewer of the chronic diseases that have too long been considered the “normal” baggage of growing old.
Literature
[1] Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A New Immune–Metabolic Viewpoint for Age-Related Diseases. Nat Rev Endocrinol 2018, 14, 576–590, doi:10.1038/S41574-018-0059-4.
[2] Ferrucci, L.; Fabbri, E. Inflammageing: Chronic Inflammation in Ageing, Cardiovascular Disease, and Frailty. Nat Rev Cardiol 2018, 15, 505–522.
[3] Qin, P.; Ho, F.K.; Celis-Morales, C.A.; Pell, J.P. Association between Systemic Inflammation Biomarkers and Incident Cardiovascular Disease in 423,701 Individuals: Evidence from the UK Biobank Cohort. Cardiovasc Diabetol 2025, 24, 162, doi:10.1186/s12933-025-02721-9.
[4] Tork, M.A.B.; Fotouhi, S.; Roozi, P.; Negah, S.S. Targeting NLRP3 Inflammasomes: A Trojan Horse Strategy for Intervention in Neurological Disorders. Mol Neurobiol 2024.
[5] López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194, doi:10.1016/J.CELL.2013.05.039.
[6] López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278, doi:10.1016/J.CELL.2022.11.001.
[7] Poole, C.S.; Allen, I.C. NF-ΚB-Inducing Kinase (NIK): An Emerging Therapeutic Target in Human Disease. Expert Opin Ther Targets 2025.
[8] Kannan, G.; Paul, B.M.; Thangaraj, P. Stimulation, Regulation, and Inflammaging Interventions of Natural Compounds on Nuclear Factor Kappa B (NF-KB) Pathway: A Comprehensive Review. Inflammopharmacology 2025.
[9] Zhang, B.; Xu, P.; Ablasser, A. Regulation of the CGAS-STING Pathway. Annu Rev Immunol 2025, 43, 667–692, doi:10.1146/annurev-immunol-101721-032910.
[10] Iskandar, M.; Xiao Barbero, M.; Jaber, M.; Chen, R.; Gomez-Guevara, R.; Cruz, E.; Westerheide, S. A Review of Telomere Attrition in Cancer and Aging: Current Molecular Insights and Future Therapeutic Approaches. Cancers (Basel) 2025, 17.
[11] Rovillain, E.; Mansfield, L.; Caetano, C.; Alvarez-Fernandez, M.; Caballero, O.L.; Medema, R.H.; Hummerich, H.; Jat, P.S. Activation of Nuclear Factor-Kappa B Signalling Promotes Cellular Senescence. Oncogene 2011, 30, 2356–2366, doi:10.1038/onc.2010.611.
[12] Lingappan, K. NF-ΚB in Oxidative Stress. Curr Opin Toxicol 2018, 7, 81–86.
[13] Tylutka, A.; Walas, Ł.; Zembron-Lacny, A. Level of IL-6, TNF, and IL-1β and Age-Related Diseases: A Systematic Review and Meta-Analysis. Front Immunol 2024, 15.
[14] Baechle, J.J.; Chen, N.; Makhijani, P.; Winer, S.; Furman, D.; Winer, D.A. Chronic Inflammation and the Hallmarks of Aging. Mol Metab 2023, 74.
[15] Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of Innate Immunity System during Aging: NF-KB Signaling Is the Molecular Culprit of Inflamm-Aging. Ageing Res Rev 2008, 7, 83–105.
[16] Xu, J.; Núñez, G. The NLRP3 Inflammasome: Activation and Regulation. Trends Biochem Sci 2023, 48, 331–344.
[17] Dadkhah, M.; Sharifi, M. The NLRP3 Inflammasome: Mechanisms of Activation, Regulation, and Role in Diseases. Int Rev Immunol 2024.
[18] Liang, R.; Qi, X.; Cai, Q.; Niu, L.; Huang, X.; Zhang, D.; Ling, J.; Wu, Y.; Chen, Y.; Yang, P.; et al. The Role of NLRP3 Inflammasome in Aging and Age-Related Diseases. Immunity and Ageing 2024, 21.
[19] NLRP3 Knockout Extends Maximum Life Span by 29% in Mice.
[20] Marín-Aguilar, F.; Lechuga-Vieco, A. V.; Alcocer-Gómez, E.; Castejón-Vega, B.; Lucas, J.; Garrido, C.; Peralta-Garcia, A.; Pérez-Pulido, A.J.; Varela-López, A.; Quiles, J.L.; et al. NLRP3 Inflammasome Suppression Improves Longevity and Prevents Cardiac Aging in Male Mice. Aging Cell 2020, 19, doi:10.1111/acel.13050.
[21] Cui, H.; Xie, L.; Lu, H.; Cheng, C.; Xue, F.; Wu, Z.; Liu, L.; Qiao, L.; Zhang, C.; Zhang, W.; et al. Macrophage Junctional Adhesion Molecule-like (JAML) Protein Promotes NLRP3 Inflammasome Activation in the Development of Atherosclerosis. Cell Death Differ 2025, doi:10.1038/s41418-025-01489-5.
[22] Mandal, S.; Ramesh, M.; Govindaraju, T. Strategic Mutations in Designer Native Peptides Combat NLRP3 Inflammasome Activation in Neurodegenerative Disorders. J Med Chem 2025, doi:10.1021/acs.jmedchem.4c02158.
[23] Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxid Med Cell Longev 2016, 2016.
[24] Olivieri, F.; Prattichizzo, F.; Grillari, J.; Balistreri, C.R. Cellular Senescence and Inflammaging in Age-Related Diseases. Mediators Inflamm 2018, 2018.
[25] Birch, J.; Gil, J. Senescence and the SASP: Many Therapeutic Avenues. Genes Dev 2020, 34, 1565–1576, doi:10.1101/gad.343129.120.
[26] Walker, K.A.; Basisty, N.; Wilson, D.M.; Ferrucci, L. Connecting Aging Biology and Inflammation in the Omics Era. Journal of Clinical Investigation 2022, 132.
[27] Paulo, F.L.; Mikolaj, S.; Olena, O.; Julien, K.; Miwa, S.; Cameron, K.; Ishaq, A.; Saretzki, G.; Nagaraja, S.; Glyn, G.; et al. The Bystander Effect Contributes to the Accumulation of Senescent Cells in Vivo. 2019, doi:10.1111/acel.12848.
[28] Liu, H.; Huang, Y.; Lyu, Y.; Dai, W.; Tong, Y.; Li, Y. GDF15 as a Biomarker of Ageing. Exp Gerontol 2021, 146, doi:10.1016/j.exger.2021.111228.
[29] Machado-Oliveira, G.; Ramos, C.; Marques, A.R.A.; Vieira, O. V. Cell Senescence, Multiple Organelle Dysfunction and Atherosclerosis. Cells 2020, 9, 1–31.
[30] Van Deursen, J.M. The Role of Senescent Cells in Ageing. Nature 2014 509:7501 2014, 509, 439–446, doi:10.1038/nature13193.
[31] Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular Senescence and Senolytics: The Path to the Clinic. Nature Medicine 2022 28:8 2022, 28, 1556–1568, doi:10.1038/s41591-022-01923-y.
[32] Fulop, T.; Larbi, A.; Dupuis, G.; Page, A. Le; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging as Two Sides of the Same Coin: Friends or Foes? Front Immunol 2018, 8.
[33] Müller, L.; Di Benedetto, S.; Pawelec, G. The Immune System and Its Dysregulation with Aging. In Subcellular Biochemistry; Springer New York, 2019; Vol. 91, pp. 21–43.
[34] Poulios, P.; Skampouras, S.; Piperi, C. Deciphering the Role of Cytokines in Aging: Biomarker Potential and Effective Targeting. Mech Ageing Dev 2025, 224, doi:10.1016/j.mad.2025.112036.
[35] Camell, C.D. Taa Cells and Granzyme K: Old Players with New Tricks. Immunity 2021, 54, 6–8, doi:10.1016/j.immuni.2020.12.008.
[36] Minato, N.; Hattori, M.; Hamazaki, Y. Physiology and Pathology of T-Cell Aging. Int Immunol 2020, 32, 223–231.
[37] Davitt, E.; Davitt, C.; Mazer, M.B.; Areti, S.S.; Hotchkiss, R.S.; Remy, K.E. COVID-19 Disease and Immune Dysregulation. Best Pract Res Clin Haematol 2022, 35.
[38] Sumbul-Sekerci, B.; Pasin, O.; Balkan, E.; Sekerci, A. The Role of Inflammation, Oxidative Stress, Neuronal Damage, and Endothelial Dysfunction in the Neuropathology of Cognitive Complications in Diabetes: A Moderation and Mediation Analysis. Brain Behav 2025, 15, doi:10.1002/brb3.70225.
[39] Torres, E.M.; Tellechea, M.L. Biomarkers of Endothelial Dysfunction and Cytokine Levels in Hypothyroidism: A Series of Meta-Analyses. Expert Rev Endocrinol Metab 2024.
[40] Zeng, Y.; Buonfiglio, F.; Li, J.; Pfeiffer, N.; Gericke, A. Mechanisms Underlying Vascular Inflammaging: Current Insights and Potential Treatment Approaches. Aging Dis 2025, doi:10.14336/AD.2024.0922.
[41] López, B.; Ravassa, S.; Moreno, M.U.; José, G.S.; Beaumont, J.; González, A.; Díez, J. Diffuse Myocardial Fibrosis: Mechanisms, Diagnosis and Therapeutic Approaches. Nat Rev Cardiol 2021, 18, 479–498.
[42] Papi, S.; Ahmadizar, F.; Hasanvand, A. The Role of Nitric Oxide in Inflammation and Oxidative Stress. Immunopathologia Persa 2019, 5.
[43] Gocmez, S.S.; Yazir, Y.; Gacar, G.; Demirtaş Şahin, T.; Arkan, S.; Karson, A.; Utkan, T. Etanercept Improves Aging-Induced Cognitive Deficits by Reducing Inflammation and Vascular Dysfunction in Rats. Physiol Behav 2020, 224, 113019, doi:10.1016/J.PHYSBEH.2020.113019.
[44] Sciorati, C.; Gamberale, R.; Monno, A.; Citterio, L.; Lanzani, C.; De Lorenzo, R.; Ramirez, G.A.; Esposito, A.; Manunta, P.; Manfredi, A.A.; et al. Pharmacological Blockade of TNFα Prevents Sarcopenia and Prolongs Survival in Aging Mice. Aging 2020, 12, 23497–23508, doi:10.18632/AGING.202200.
[45] Chung, J.Y.; Kim, S.G.; Kim, S.H.; Park, C.H. Sarcopenia: How to Determine and Manage. Knee Surg Relat Res 2025, 37.
[46] Liu, L.; Shi, Z.; Ji, X.; Zhang, W.; Luan, J.; Zahr, T.; Qiang, L. Adipokines, Adiposity, and Atherosclerosis. Cellular and Molecular Life Sciences 2022, 79, doi:10.1007/S00018-022-04286-2.
[47] Krueger, A.B.C.; Zhu, X.; Siddiqi, S.; Whitehead, E.C.; Tang, H.; Jordan, K.L.; Lerman, A.; Lerman, L.O. Mesenchymal Stem/Stromal Cells Reverse Adipose Tissue Inflammation in Pigs with Metabolic Syndrome and Renovascular Hypertension. Cells 2025, 14, doi:10.3390/cells14010040.
[48] Sayiner, M.; Koenig, A.; Henry, L.; Younossi, Z.M. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in the United States and the Rest of the World. Clin Liver Dis 2016, 20, 205–214.
[49] Niewerth, D.; Kaspers, G.J.; Assaraf, Y.G.; Van Meerloo, J.; Kirk, C.J.; Anderl, J.; Blank, J.L.; Van De Ven, P.M.; Zweegman, S.; Jansen, G.; et al. Interferon-γ-Induced Upregulation of Immunoproteasome Subunit Assembly Overcomes Bortezomib Resistance in Human Hematological Cell Lines. J Hematol Oncol 2014, 7, doi:10.1186/1756-8722-7-7.
[50] Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R.C. IL-1β, IL-6, TNF- α and CRP in Elderly Patients with Depression or Alzheimer’s Disease: Systematic Review and Meta-Analysis. Sci Rep 2018, 8, doi:10.1038/s41598-018-30487-6.
[51] Mallick, R.; Basak, S.; Chowdhury, P.; Bhowmik, P.; Das, R.K.; Banerjee, A.; Paul, S.; Pathak, S.; Duttaroy, A.K. Targeting Cytokine-Mediated Inflammation in Brain Disorders: Developing New Treatment Strategies. Pharmaceuticals (Basel) 2025, 18, doi:10.3390/ph18010104.
[52] Qin, Q.; Teng, Z.; Liu, C.; Li, Q.; Yin, Y.; Tang, Y. TREM2, Microglia, and Alzheimer’s Disease. Mech Ageing Dev 2021, 195, 111438, doi:10.1016/j.mad.2021.111438.
[53] Rivers-Auty, J.; Mather, A.E.; Peters, R.; Lawrence, C.B.; Brough, D. Anti-Inflammatories in Alzheimer’s Disease-Potential Therapy or Spurious Correlate? Brain Commun 2020, 2, doi:10.1093/braincomms/fcaa109.
[54] Qian, Y.; Wang, X.; Zhu, C.; Tong, Q.; Zhang, J.; Zhao, S. Causal Association Between Chronic Inflammatory Arthropathies and Sarcopenia and the Mediation Role of Inflammatory Factors. Int J Rheum Dis 2025, 28, doi:10.1111/1756-185X.70100.
[55] Livshits, G.; Kalinkovich, A. Targeting Chronic Inflammation as a Potential Adjuvant Therapy for Osteoporosis. Life Sci 2022, 306.
[56] Visconti, L.; Santoro, D.; Cernaro, V.; Buemi, M.; Lacquaniti, A. Kidney-Lung Connections in Acute and Chronic Diseases: Current Perspectives. J Nephrol 2016, 29, 341–348.
[57] Liu, W.; Zhang, X.; Chen, X. Unraveling the Causal Associations between Systemic Cytokines and Six Inflammatory Skin Diseases. Cytokine 2025, 185, doi:10.1016/j.cyto.2024.156810.
[58] Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and Anti-Inflammaging: A Systemic Perspective on Aging and Longevity Emerged from Studies in Humans. Mech. Ageing Dev. 2007, 128, 92–105, doi:10.1016/j.mad.2006.11.016.
[59] Joseph, P.; Lanas, F.; Roth, G.; Lopez-Jaramillo, P.; Lonn, E.; Miller, V.; Mente, A.; Leong, D.; Schwalm, J.D.; Yusuf, S. Cardiovascular Disease in the Americas: The Epidemiology of Cardiovascular Disease and Its Risk Factors. The Lancet Regional Health – Americas 2025, 42.
[60] Aday, A.W.; Ridker, P.M. Antiinflammatory Therapy in Clinical Care: The CANTOS Trial and Beyond. Front Cardiovasc Med 2018, 5.
[61] Orkaby, A.R.; Thomson, A.; MacFadyen, J.; Besdine, R.; Forman, D.E.; Travison, T.G.; Ridker, P.M. Effect of Canakinumab on Frailty: A Post Hoc Analysis of the CANTOS Trial. Aging Cell 2024, 23, doi:10.1111/acel.14029.
[62] Woo, J.; Zhai, T.; Yang, F.; Xu, H.; Healey, M.L.; Yates, D.P.; Beste, M.T.; Steensma, D.P. Effect of Clonal Hematopoiesis Mutations and Canakinumab Treatment on Incidence of Solid Tumors in the CANTOS Randomized Clinical Trial. Cancer Prevention Research 2024, 17, 429–436, doi:10.1158/1940-6207.CAPR-23-0342.
[63] Luo, Y.; Yang, L.; Cheng, X.J.; Bai, Y.P.; Xiao, Z.L. The Association between Blood Count Based Inflammatory Markers and the Risk of Atrial Fibrillation Heart Failure and Cardiovascular Mortality. Sci Rep 2025, 15, doi:10.1038/s41598-025-94507-y.
[64] Mangoni, A.A.; Wiese, M.D.; Woodman, R.J.; Sotgia, S.; Zinellu, A.; Carru, C.; Hulin, J.A.; Shanahan, E.M.; Tommasi, S. Methotrexate, Blood Pressure and Arterial Function in Rheumatoid Arthritis: Study Protocol. Future Cardiol 2024, doi:10.1080/14796678.2024.2411167.
[65] Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease:
[66] Haines, D.D.; Trushin, M. V.; Rose, S.; Bernard, I.A.S.; Mahmoud, F.F. Parkinson’s Disease: Alpha Synuclein, Heme Oxygenase and Biotherapeutic Countermeasures. Curr Pharm Des 2018, 24, 2317–2321, doi:10.2174/1381612824666180717161338.
[67] Qin, Q.; Wang, D.; Qu, Y.; Li, J.; An, K.; Mao, Z.; Li, J.; Xiong, Y.; Min, Z.; Xue, Z. Enhanced Glycolysis-Derived Lactate Promotes Microglial Activation in Parkinson’s Disease via Histone Lactylation. NPJ Parkinsons Dis 2025, 11, doi:10.1038/s41531-024-00858-0.
[68] Yamamoto-Furusho, J.K.; Gutierrez-Herrera, F.D. Molecular Mechanisms and Clinical Aspects of Colitis-Associated Cancer in Ulcerative Colitis. Cells 2025, 14.
[69] Granel, N.; Iserte, G.; Bartres, C.; Llarch, N.; Pla, A.; Sapena, V.; Mariño, Z.; Lens, S.; Vilana, R.; Nuñez, I.; et al. Liver Cancer Risk and Changes in Lifestyle Habits after Successful Hepatitis C Virus Therapy Post-DAA HCV Therapy: Lifestyle Changes and Liver Cancer Risk. BMC Gastroenterol 2025, 25, doi:10.1186/s12876-025-03611-w.
[70] Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov 2022, 12, 31–46.
[71] Cui, Y.; Cui, S.; Lu, W.; Wang, Y.; Zhuo, Z.; Wang, R.; Zhang, D.; Wu, X.; Chang, L.; Zuo, X.; et al. CRP, IL-1α, IL-1β, and IL-6 Levels and the Risk of Breast Cancer: A Two-Sample Mendelian Randomization Study. Sci Rep 2024, 14, doi:10.1038/s41598-024-52080-w.
[72] Morrison, W.B. Inflammation and Cancer: A Comparative View. J Vet Intern Med 2012, 26, 18–31.
[73] Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-Induced DNA Damage, Mutations and Cancer. DNA Repair (Amst) 2019, 83, 102673, doi:10.1016/J.DNAREP.2019.102673.
[74] Zhao, Y.; Simon, M.; Seluanov, A.; Gorbunova, V. DNA Damage and Repair in Age-Related Inflammation. Nat Rev Immunol 2023, 23, 75–89.
[75] Lv, Z.; Jiao, J.; Xue, W.; Shi, X.; Wang, R.; Wu, J. Activation-Induced Cytidine Deaminase in Tertiary Lymphoid Structures: Dual Roles and Implications in Cancer Prognosis. Front Oncol 2025, 15, doi:10.3389/fonc.2025.1555491.
[76] Ni, C.W.; Hsieh, H.J.; Chao, Y.J.; Wang, D.L. Interleukin-6-Induced JAK2/STAT3 Signaling Pathway in Endothelial Cells Is Suppressed by Hemodynamic Flow. Am J Physiol Cell Physiol 2004, 287, doi:10.1152/ajpcell.00532.2003.
[77] Kang, L.; Cao, J.; Guo, W.; Cui, X.; Wei, Y.; Zhang, J.; Liu, F.; Duan, C.; Lin, Q.; Lv, P.; et al. Tumor Necrosis Factor-α–Dependent Inflammation Upregulates High Mobility Group Box 1 To Induce Tumor Promotion and Anti–Programmed Cell Death Protein-1 Immunotherapy Resistance in Lung Adenocarcinoma. Laboratory Investigation 2025, 105, doi:10.1016/j.labinv.2024.102164.
[78] Desai, S.A.; Patel, V.P.; Bhosle, K.P.; Nagare, S.D.; Thombare, K.C. The Tumor Microenvironment: Shaping Cancer Progression and Treatment Response. Journal of Chemotherapy 2023.
[79] Panwar, P.; Butler, G.S.; Jamroz, A.; Azizi, P.; Overall, C.M.; Brömme, D. Aging-Associated Modifications of Collagen Affect Its Degradation by Matrix Metalloproteinases. Matrix Biology 2018, doi:10.1016/j.matbio.2017.06.004.
[80] Bielenberg, D.R.; D’Amore, P.A. Angiogenesis: Biology and Pathology, Second Edition. Cold Spring Harb Perspect Med 2024, a041779, doi:10.1101/cshperspect.a041779.
[81] Olivieri, F.; Rippo, M.R.; Monsurrò, V.; Salvioli, S.; Capri, M.; Procopio, A.D.; Franceschi, C. MicroRNAs Linking Inflamm-Aging, Cellular Senescence and Cancer. Ageing Res Rev 2013, 12, 1056–1068, doi:10.1016/j.arr.2013.05.001.
[82] Huang, Y.; Che, X.; Wang, P.W.; Qu, X. P53/MDM2 Signaling Pathway in Aging, Senescence and Tumorigenesis. Semin Cancer Biol 2024, 101, 44–57.
[83] Wong, C.C.; Baum, J.; Silvestro, A.; Beste, M.T.; Bharani-Dharan, B.; Xu, S.; Wang, Y.A.; Wang, X.; Prescott, M.F.; Krajkovich, L.; et al. Inhibition of IL1β by Canakinumab May Be Effective against Diverse Molecular Subtypes of Lung Cancer: An Exploratory Analysis of the CANTOS Trial AC. Cancer Res 2020, 80, 5597–5605, doi:10.1158/0008-5472.CAN-19-3176.
[84] Viana-Huete, V.; Fuster, J.J. Potential Therapeutic Value of Interleukin 1b-Targeted Strategies in Atherosclerotic Cardiovascular Disease. Revista Española de Cardiología (English Edition) 2019, 72, 760–766, doi:10.1016/j.rec.2019.03.006.
[85] Orchard, S.G.; Lockery, J.E.; Broder, J.C.; Ernst, M.E.; Espinoza, S.; Gibbs, P.; Wolfe, R.; Polekhina, G.; Zoungas, S.; Loomans-Kropp, H.A.; et al. Association of Metformin, Aspirin, and Cancer Incidence with Mortality Risk in Adults with Diabetes. JNCI Cancer Spectr 2023, 7, doi:10.1093/jncics/pkad017.
[86] Driscoll, C.B.; Rich, J.M.; Isaacson, D.; Nicolas, J.; Jiang, Y.; Mi, X.; Yang, C.; Kocsuta, V.; Goh, R.; Patel, N.; et al. Tumor Necrosis Factor-Alpha Inhibitor Use and Malignancy Risk: A Systematic Review and Patient Level Meta-Analysis. Cancers (Basel) 2025, 17.
[87] Avouac, J.; Ait-Oufella, H.; Habauzit, C.; Benkhalifa, S.; Combe, B. The Cardiovascular Safety of Tumour Necrosis Factor Inhibitors in Arthritic Conditions: A Structured Review with Recommendations. Rheumatol Ther 2025.
[88] Li, J.; Zhang, Z.; Wu, X.; Zhou, J.; Meng, D.; Zhu, P. Risk of Adverse Events After Anti-TNF Treatment for Inflammatory Rheumatological Disease. A Meta-Analysis. Front Pharmacol 2021, 12.
[89] Cavalli, G.; De Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Tassan Din, C.; Boffini, N.; et al. Interleukin-1 Blockade with High-Dose Anakinra in Patients with COVID-19, Acute Respiratory Distress Syndrome, and Hyperinflammation: A Retrospective Cohort Study. Lancet Rheumatol 2020, 2, e325–e331, doi:10.1016/S2665-9913(20)30127-2.
[90] Singh, J.A.; Saba, B.; Lopez-Olivo, M.A. Tocilizumab for Rheumatoid Arthritis: A Cochrane Systematic Review. Journal of Rheumatology 2011, 38, 10–20.
[91] Marcum, Z.A.; Hanlon, J.T. Recognizing the Risks of Chronic Nonsteroidal Anti-Inflammatory Drug Use in Older Adults. Annals of Long-Term Care 2010, 18, 24–27.
[92] Stanbury, R.M.; Graham, E.M. Systemic Corticosteroid Therapy – Side Effects and Their Management. British Journal of Ophthalmology 1998, 82, 704–708.
[93] Cleland, J.G.F. Aspirin for Secondary Prevention of Atherosclerosis-Evidence or Dogma? JAMA Cardiol 2025, 10, 114–116, doi:10.1001/jamacardio.2024.4335.
[94] Diamantis, E.; Kyriakos, G.; Quiles-Sanchez, L.V.; Farmaki, P.; Troupis, T. The Anti-Inflammatory Effects of Statins on Coronary Artery Disease: An Updated Review of the Literature. Curr Cardiol Rev 2017, 13, doi:10.2174/1573403×13666170426104611.
[95] Sparks, J.A.; Barbhaiya, M.; Karlson, E.W.; Ritter, S.Y.; Raychaudhuri, S.; Corrigan, C.C.; Lu, F.; Selhub, J.; Chasman, D.I.; Paynter, N.P.; et al. Investigating Methotrexate Toxicity within a Randomized Double-Blinded, Placebo-Controlled Trial: Rationale and Design of the Cardiovascular Inflammation Reduction Trial-Adverse Events (CIRT-AE) Study. Semin Arthritis Rheum 2017, 47, 133–142, doi:10.1016/j.semarthrit.2017.02.003.
[96] Lamberg, O.; Pandher, K.; Troost, J.P.; Lim, H.W. Long-Term Adverse Event Risks of Oral JAK Inhibitors versus Immunomodulators: A Literature Review. Arch Dermatol Res 2025, 317.
[97] Zheng, Y.; Zhang, X.; Wang, Z.; Zhang, R.; Wei, H.; Yan, X.; Jiang, X.; Yang, L. MCC950 as a Promising Candidate for Blocking NLRP3 Inflammasome Activation: A Review of Preclinical Research and Future Directions. Arch Pharm (Weinheim) 2024.
[98] Mezzaroma, E.; Abbate, A.; Toldo, S. NLRP3 Inflammasome Inhibitors in Cardiovascular Diseases. Molecules 2021, 26.
[99] Li, Z.; Gong, C. NLRP3 Inflammasome in Alzheimer’s Disease: Molecular Mechanisms and Emerging Therapies. Front Immunol 2025, 16, doi:10.3389/fimmu.2025.1583886.
[100] Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V. V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination Inactivates Gasdermin D and Blocks Pyroptosis. Science (1979) 2020, 369, doi:10.1126/science.abb9818.
[101] Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The CGAS–STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat Rev Immunol 2021, 21, 548–569.
[102] Bailey, K.L.; Smith, L.M.; Heires, A.J.; Katafiasz, D.M.; Romberger, D.J.; LeVan, T.D. Aging Leads to Dysfunctional Innate Immune Responses to TLR2 and TLR4 Agonists. Aging Clin Exp Res 2019, 31, 1185–1193, doi:10.1007/s40520-018-1064-0.
[103] Xu, Y.; Bai, L.; Yang, X.; Huang, J.; Wang, J.; Wu, X.; Shi, J. Recent Advances in Anti-Inflammation via AMPK Activation. Heliyon 2024, 10.
[104] Wang, M.; Zhou, S.; Hu, Y.; Tong, W.; Zhou, H.; Ma, M.; Cai, X.; Zhang, Z.; Zhang, L.; Chen, Y. Macrophages Overexpressing Interleukin-10 Target and Prevent Atherosclerosis: Regression of Plaque Formation and Reduction in Necrotic Core. Bioeng Transl Med 2024, doi:10.1002/btm2.10717.
[105] Islam, M.T.; Tuday, E.; Allen, S.; Kim, J.; Trott, D.W.; Holland, W.L.; Donato, A.J.; Lesniewski, L.A. Senolytic Drugs, Dasatinib and Quercetin, Attenuate Adipose Tissue Inflammation, and Ameliorate Metabolic Function in Old Age. Aging Cell 2023, 22, e13767, doi:10.1111/ACEL.13767.
[106] Spiegel, M. Fisetin as a Blueprint for Senotherapeutic Agents – Elucidating Geroprotective and Senolytic Properties with Molecular Modeling. Chemistry – A European Journal 2025, doi:10.1002/chem.202403755.
[107] Watson, M.M.; van der Giezen, M.; Søreide, K. Gut Microbiome Influence on Human Epigenetics, Health, and Disease. Handbook of Epigenetics: The New Molecular and Medical Genetics, Third Edition 2023, 669–686, doi:10.1016/B978-0-323-91909-8.00012-8.
[108] Hung, T. Van; Suzuki, T. Dietary Fermentable Fibers Attenuate Chronic Kidney Disease in Mice by Protecting the Intestinal Barrier. J Nutr 2018, 148, 552–561, doi:10.1093/JN/NXY008.
[109] Ahmed, M.; Riaz, U.; Lv, H.; Amjad, M.; Ahmed, S.; Ali, S.; Ghani, M.U.; Hua, G.; Yang, L. Nicotinamide Mononucleotide Restores NAD+ Levels to Alleviate LPS-Induced Inflammation via the TLR4/NF-ΚB/MAPK Signaling Pathway in Mice Granulosa Cells. Antioxidants 2025, 14, doi:10.3390/antiox14010039.
[110] Walker, M.A.; Tian, R. NAD(H) in Mitochondrial Energy Transduction: Implications for Health and Disease. Curr Opin Physiol 2018, 3, 101–109.
[111] van Nostrand, J.L.; Hellberg, K.; Luo, E.C.; van Nostrand, E.L.; Dayn, A.; Yu, J.; Shokhirev, M.N.; Dayn, Y.; Yeo, G.W.; Shaw, R.J. AMPK Regulation of Raptor and TSC2 Mediate Metformin Effects on Transcriptional Control of Anabolism and Inflammation. Genes Dev 2020, 34, 1330–1344, doi:10.1101/GAD.339895.120.
[112] Mathur, N.; Pedersen, B.K. Exercise as a Mean to Control Low-Grade Systemic Inflammation. Mediators Inflamm 2008, 2008.
[113] Chen, C.; Zhang, D.; Ye, M.; You, Y.; Song, Y.; Chen, X. Effects of Various Exercise Types on Inflammatory Response in Individuals with Overweight and Obesity: A Systematic Review and Network Meta-Analysis of Randomized Controlled Trials. Int J Obes 2025, 49, 214–225, doi:10.1038/s41366-024-01649-6.
[114] Cesari, M.; Vellas, B.; Hsu, F.C.; Newman, A.B.; Doss, H.; King, A.C.; Manini, T.M.; Church, T.; Gill, T.M.; Miller, M.E.; et al. A Physical Activity Intervention to Treat the Frailty Syndrome in Older Persons – Results from the LIFE-P Study. Journals of Gerontology – Series A Biological Sciences and Medical Sciences 2015, 70, 216–222, doi:10.1093/gerona/glu099.
[115] Takahashi, H.; Alves, C.R.R.; Stanford, K.I.; Middelbeek, R.J.W.; Nigro, P.; Ryan, R.E.; Xue, R.; Sakaguchi, M.; Lynes, M.D.; So, K.; et al. TGF-Β2 Is an Exercise-Induced Adipokine That Regulates Glucose and Fatty Acid Metabolism. Nature Metabolism 2019 1:2 2019, 1, 291–303, doi:10.1038/s42255-018-0030-7.
[116] Guo, Q.; Luo, Q.; Song, G. Control of Muscle Satellite Cell Function by Specific Exercise-Induced Cytokines and Their Applications in Muscle Maintenance. J Cachexia Sarcopenia Muscle 2024, 15, 466–476, doi:10.1002/JCSM.13440.
[117] Lavin, K.M.; Perkins, R.K.; Jemiolo, B.; Raue, U.; Trappe, S.W.; Trappe, T.A. Effects of Aging and Lifelong Aerobic Exercise on Basal and Exercise-Induced Inflammation. J Appl Physiol (1985) 2020, 128, 87–99, doi:10.1152/japplphysiol.00495.2019.
[118] Rech, A.; Botton, C.E.; Lopez, P.; Quincozes-Santos, A.; Umpierre, D.; Pinto, R.S. Effects of Short-Term Resistance Training on Endothelial Function and Inflammation Markers in Elderly Patients with Type 2 Diabetes: A Randomized Controlled Trial. Exp Gerontol 2019, 118, 19–25, doi:10.1016/j.exger.2019.01.003.
[119] American Heart Association Recommendations for Physical Activity in Adults and Kids.
[120] Ifland, J.R.; Preuss, H.G.; Marcus, M.T.; Rourke, K.M.; Taylor, W.C.; Wright, H.T.; Sheppard, K.K. Functional Foods in the Treatment of Processed Food Addiction and the Metabolic Syndrome. Nutraceuticals and Functional Foods in Human Health and Disease Prevention 2015, 43–60, doi:10.1201/B19308/NUTRACEUTICALS-FUNCTIONAL-FOODS-HUMAN-HEALTH-DISEASE-PREVENTION-HARRY-PREUSS-DEBASIS-BAGCHI-ANAND-SWAROOP.
[121] Liang, S.; Zhou, Y.; Zhang, Q.; Yu, S.; Wu, S. Ultra-Processed Foods and Risk of All-Cause Mortality: An Updated Systematic Review and Dose-Response Meta-Analysis of Prospective Cohort Studies. Syst Rev 2025, 14.
[122] Scarmeas, N.; Stern, Y.; Tang, M.; Luchsinger, J.A. Mediterranean Diet and Risk for Alzheimer’s Disease Nikolaos. Ann Neurol 2011, 59, 912–921, doi:10.1002/ana.20854.Mediterranean.
[123] Razquin, C.; Martinez-Gonzalez, M.A. A Traditional Mediterranean Diet Effeectively Reduces Inflammation and Improves Cardiovascular Health. Nutrients 2019, 11.
[124] Carluccio, M.A.; Siculella, L.; Ancora, M.A.; Massaro, M.; Scoditti, E.; Storelli, C.; Visioli, F.; Distante, A.; De Caterina, R. Olive Oil and Red Wine Antioxidant Polyphenols Inhibit Endothelial Activation. Arterioscler Thromb Vasc Biol 2003, 23, 622–629, doi:10.1161/01.ATV.0000062884.69432.A0.
[125] Nicklas, B.J.; You, T.; Pahor, M. Behavioural Treatments for Chronic Systemic Inflammation: Effects of Dietary Weight Loss and Exercise Training. CMAJ. Canadian Medical Association Journal 2005, 172, 1199–1209.
[126] Calder, P.C.; Bosco, N.; Bourdet-Sicard, R.; Capuron, L.; Delzenne, N.; Doré, J.; Franceschi, C.; Lehtinen, M.J.; Recker, T.; Salvioli, S.; et al. Health Relevance of the Modification of Low Grade Inflammation in Ageing (Inflammageing) and the Role of Nutrition. Ageing Res Rev 2017, 40, 95–119.
[127] Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867.
[128] Taormina, G.; Mirisola, M.G. Calorie Restriction in Mammals and Simple Model Organisms. Biomed Res Int 2014, 2014.
[129] Haasis, E.; Bettenburg, A.; Lorentz, A. Effect of Intermittent Fasting on Immune Parameters and Intestinal Inflammation. Nutrients 2024, 16, doi:10.3390/nu16223956.
[130] Larson-Meyer, D.E.; Newcomer, B.R.; Heilbronn, L.K.; Volaufova, J.; Smith, S.R.; Alfonso, A.J.; Lefevre, M.; Rood, J.C.; Williamson, D.A.; Ravussin, E.; et al. Effect of 6-Month Calorie Restriction and Exercise on Serum and Liver Lipids and Markers of Liver Function. Obesity (Silver Spring) 2008, 16, 1355–1362, doi:10.1038/oby.2008.201.
[131] Meydani, S.N.; Das, S.K.; Pieper, C.F.; Lewis, M.R.; Klein, S.; Dixit, V.D.; Gupta, A.K.; Villareal, D.T.; Bhapkar, M.; Huang, M.; et al. Long-Term Moderate Calorie Restriction Inhibits Inflammation without Impairing Cell-Mediated Immunity: A Randomized Controlled Trial in Non-Obese Humans. Aging 2016, 8, 1416–1431, doi:10.18632/aging.100994.
[132] Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat Rev Mol Cell Biol 2018, 19, 121–135, doi:10.1038/nrm.2017.95.
[133] Salminen, A.; Kaarniranta, K.; Kauppinen, A. Age-Related Changes in AMPK Activation: Role for AMPK Phosphatases and Inhibitory Phosphorylation by Upstream Signaling Pathways. Ageing Res Rev 2016, 28, 15–26, doi:10.1016/j.arr.2016.04.003.
[134] Black, D.S.; Slavich, G.M. Mindfulness Meditation and the Immune System: A Systematic Review of Randomized Controlled Trials. Ann N Y Acad Sci 2016, 1373, 13–24, doi:10.1111/NYAS.12998.
[135] Rusch, H.L.; Rosario, M.; Levison, L.M.; Olivera, A.; Livingston, W.S.; Wu, T.; Gill, J.M. The Effect of Mindfulness Meditation on Sleep Quality: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Ann N Y Acad Sci 2019, 1445, 5–16, doi:10.1111/NYAS.13996.
[136] Hammami, S.; Lizard, G.; Vejux, A.; Deb, S.; Berei, J.; Miliavski, E.; Khan, M.J.; Broder, T.J.; Akurugo, T.A.; Lund, C.; et al. The Effects of Smoking on Telomere Length, Induction of Oncogenic Stress, and Chronic Inflammatory Responses Leading to Aging. Cells 2024, Vol. 13, Page 884 2024, 13, 884, doi:10.3390/CELLS13110884.
[137] Imhof, A.; Froehlich, M.; Brenner, H.; Boeing, H.; Pepys, M.B.; Koenig, W. Effect of Alcohol Consumption on Systemic Markers of Inflammation. Lancet 2001, 357, 763–767, doi:10.1016/S0140-6736(00)04170-2.
[138] van ’t Klooster, C.C.; van der Graaf, Y.; Ridker, P.M.; Westerink, J.; Hjortnaes, J.; Sluijs, I.; Asselbergs, F.W.; Bots, M.L.; Kappelle, L.J.; Visseren, F.L.J. The Relation between Healthy Lifestyle Changes and Decrease in Systemic Inflammation in Patients with Stable Cardiovascular Disease. Atherosclerosis 2020, 301, 37–43, doi:10.1016/j.atherosclerosis.2020.03.022.
[139] Baechle, J.J.; Chen, N.; Makhijani, P.; Winer, S.; Furman, D.; Winer, D.A. Chronic Inflammation and the Hallmarks of Aging. Mol Metab 2023, 74.
[140] Yu, W.; Tu, Y.; Long, Z.; Liu, J.; Kong, D.; Peng, J.; Wu, H.; Zheng, G.; Zhao, J.; Chen, Y.; et al. Reactive Oxygen Species Bridge the Gap between Chronic Inflammation and Tumor Development. Oxid Med Cell Longev 2022, 2022.
[141] Çakan, E.; Gunaydin, G. Activation Induced Cytidine Deaminase: An Old Friend with New Faces. Front Immunol 2022, 13.
[142] McCool, K.W.; Miyamoto, S. DNA Damage-Dependent NF-ΚB Activation: NEMO Turns Nuclear Signaling inside Out. Immunol Rev 2012, 246, 311–326, doi:10.1111/j.1600-065X.2012.01101.x.
[143] O’Donovan, A.; Pantell, M.S.; Puterman, E.; Dhabhar, F.S.; Blackburn, E.H.; Yaffe, K.; Cawthon, R.M.; Opresko, P.L.; Hsueh, W.C.; Satterfield, S.; et al. Cumulative Inflammatory Load Is Associated with Short Leukocyte Telomere Length in the Health, Aging and Body Composition Study. PLoS One 2011, 6, doi:10.1371/journal.pone.0019687.
[144] Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The Impact of Oxidative DNA Damage and Stress on Telomere Homeostasis. Mech Ageing Dev 2019, 177, 37–45, doi:10.1016/J.MAD.2018.03.013.
[145] Zhang, J.; Ju, Z. Telomere, DNA Damage, and Oxidative Stress in Stem Cell Aging. Birth Defects Res C Embryo Today 2010, 90, 297–307, doi:10.1002/BDRC.20190.
[146] Rodier, F.; Coppé, J.P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat Cell Biol 2009, 11, 973–979, doi:10.1038/NCB1909.
[147] Matthe, D.M.; Thoma, O.M.; Sperka, T.; Neurath, M.F.; Waldner, M.J. Telomerase Deficiency Reflects Age-Associated Changes in CD4+ T Cells. Immunity and Ageing 2022, 19, doi:10.1186/s12979-022-00273-0.
[148] Switzer, C.H.; Cho, H.J.; Eykyn, T.R.; Lavender, P.; Eaton, P. NOS2 and S-Nitrosothiol Signaling Induces DNA Hypomethylation and LINE-1 Retrotransposon Expression. Proc Natl Acad Sci U S A 2022, 119, doi:10.1073/pnas.2200022119.
[149] Ligthart, S.; Marzi, C.; Aslibekyan, S.; Mendelson, M.M.; Conneely, K.N.; Tanaka, T.; Colicino, E.; Waite, L.L.; Joehanes, R.; Guan, W.; et al. DNA Methylation Signatures of Chronic Low-Grade Inflammation Are Associated with Complex Diseases. Genome Biol 2016, 17, doi:10.1186/s13059-016-1119-5.
[150] Bhatt, D.; Ghosh, S. Regulation of the NF-ΚB-Mediated Transcription of Inflammatory Genes. Front Immunol 2014, 5.
[151] Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via DsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986, doi:10.1016/j.cell.2015.07.011.
[152] Fitzgerald, K.N.; Hodges, R.; Hanes, D.; Stack, E.; Cheishvili, D.; Szyf, M.; Henkel, J.; Twedt, M.W.; Giannopoulou, D.; Herdell, J.; et al. Potential Reversal of Epigenetic Age Using a Diet and Lifestyle Intervention: A Pilot Randomized Clinical Trial. Aging 2021, 13, 9419–9432, doi:10.18632/aging.202913.
[153] Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature 2013, 493, 674–678, doi:10.1038/nature11729.
[154] Kammerl, I.E.; Flexeder, C.; Karrasch, S.; Thorand, B.; Heier, M.; Peters, A.; Schulz, H.; Meiners, S. Blood Immunoproteasome Activity Is Regulated by Sex, Age and in Chronic Inflammatory Diseases: A First Population-Based Study. Cells 2021, 10, doi:10.3390/cells10123336.
[155] Zhang, K.; Kaufman, R.J. From Endoplasmic-Reticulum Stress to the Inflammatory Response. Nature 2008, 454, 455–462.
[156] Zinecker, H.; Simon, A.K. Autophagy Takes It All – Autophagy Inducers Target Immune Aging. DMM Disease Models and Mechanisms 2022, 15, doi:10.1242/DMM.049345.
[157] Madeo, F.; Pietrocola, F.; Eisenberg, T.; Kroemer, G. Caloric Restriction Mimetics: Towards a Molecular Definition. Nat Rev Drug Discov 2014, 13, 727–740.
[158] West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557, doi:10.1038/nature14156.
[159] Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome Activation Leads to Caspase-1-Dependent Mitochondrial Damage and Block of Mitophagy. Proc Natl Acad Sci U S A 2014, 111, 15514–15519, doi:10.1073/pnas.1414859111.
[160] Koo, S. jie; Garg, N.J. Metabolic Programming of Macrophage Functions and Pathogens Control. Redox Biol 2019, 24.
[161] Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of Macrophage Metabolism and Activation by MTOR and Akt Signaling. Semin Immunol 2015, 27, 286–296.
[162] Kandhaya-Pillai, R.; Miro-Mur, F.; Alijotas-Reig, J.; Tchkonia, T.; Kirkland, J.L.; Schwartz, S. TNFα-Senescence Initiates a STAT-Dependent Positive Feedback Loop, Leading to a Sustained Interferon Signature, DNA Damage, and Cytokine Secretion. Aging 2017, 9, 2411–2435, doi:10.18632/aging.101328.
[163] Lagnado, A.; Leslie, J.; Ruchaud‐Sparagano, M.; Victorelli, S.; Hirsova, P.; Ogrodnik, M.; Collins, A.L.; Vizioli, M.G.; Habiballa, L.; Saretzki, G.; et al. Neutrophils Induce Paracrine Telomere Dysfunction and Senescence in ROS‐dependent Manner. EMBO J 2021, 40, doi:10.15252/embj.2020106048.
[164] Fu, T.E.; Zhou, Z. Senescent Cells as a Target for Anti-Aging Interventions: From Senolytics to Immune Therapies. J Transl Int Med 2025, 13, 33–47, doi:10.1515/jtim-2025-0005.
[165] Kovtonyuk, L. V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front Immunol 2016, 7, 217314, doi:10.3389/FIMMU.2016.00502/BIBTEX.
[166] Woolthuis, C.M.; Stranahan, A.W.; Park, C.Y.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Jordan, C.T. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37, doi:10.1016/j.stem.2016.06.001.
[167] Pietras, E.M.; Lakshminarasimhan, R.; Techner, J.M.; Fong, S.; Flach, J.; Binnewies, M.; Passegué, E. Re-Entry into Quiescence Protects Hematopoietic Stem Cells from the Killing Effect of Chronic Exposure to Type I Interferons. Journal of Experimental Medicine 2014, 211, 245–262, doi:10.1084/jem.20131043.
[168] Kovtonyuk, L. V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front Immunol 2016, 7.
[169] Islam, O.; Gong, X.; Rose-John, S.; Heese, K. Interleukin-6 and Neural Stem Cells: More than Gliogenesis. Mol Biol Cell 2009, 20, 188–199, doi:10.1091/mbc.E08-05-0463.
[170] Zeng, X.; Li, X.; Li, X.; Wei, C.; Shi, C.; Hu, K.; Kong, D.; Luo, Q.; Xu, Y.; Shan, W.; et al. Fecal Microbiota Transplantation from Young Mice Rejuvenates Aged Hematopoietic Stem Cells by Suppressing Inflammation. Blood 2023, 141, 1691–1707, doi:10.1182/blood.2022017514.
[171] He, H.; Wang, J. Inflammation and Hematopoietic Stem Cells Aging. Blood science (Baltimore, Md.) 2021, 3, 1–5, doi:10.1097/BS9.0000000000000063.
[172] Xing, Y.; Xuan, F.; Wang, K.; Zhang, H. Aging under Endocrine Hormone Regulation. Front Endocrinol (Lausanne) 2023, 14.
[173] Saccon, T.D.; Nagpal, R.; Yadav, H.; Cavalcante, M.B.; Nunes, A.D.D.C.; Schneider, A.; Gesing, A.; Hughes, B.; Yousefzadeh, M.; Tchkonia, T.; et al. Senolytic Combination of Dasatinib and Quercetin Alleviates Intestinal Senescence and Inflammation and Modulates the Gut Microbiome in Aged Mice. The Journals of Gerontology: Series A 2021, 76, 1895–1905, doi:10.1093/GERONA/GLAB002.
[174] Golshany, H.; Helmy, S.A.; Morsy, N.F.S.; Kamal, A.; Yu, Q.; Fan, L. The Gut Microbiome across the Lifespan: How Diet Modulates Our Microbial Ecosystem from Infancy to the Elderly. Int J Food Sci Nutr 2024.
[175] Bana, B.; Cabreiro, F. The Microbiome and Aging. Annu Rev Genet 2019, 53, 239–261.
[176] Zeng, Y.; Buonfiglio, F.; Li, J.; Pfeiffer, N.; Gericke, A. Mechanisms Underlying Vascular Inflammaging: Current Insights and Potential Treatment Approaches. Aging Dis 2025, 0, doi:10.14336/AD.2024.0922.
[177] Xie, H.; Wei, L.; Ruan, G.; Zhang, H.; Shi, H. Inflammaging Score as a Potential Prognostic Tool for Cancer: A Population-Based Cohort Study. Mech Ageing Dev 2024, 219, doi:10.1016/j.mad.2024.111939.
[178] Sayed, N.; Huang, Y.; Nguyen, K.; Krejciova-Rajaniemi, Z.; Grawe, A.P.; Gao, T.; Tibshirani, R.; Hastie, T.; Alpert, A.; Cui, L.; et al. An Inflammatory Aging Clock (IAge) Based on Deep Learning Tracks Multimorbidity, Immunosenescence, Frailty and Cardiovascular Aging. Nat Aging 2021, 1, 598–615, doi:10.1038/s43587-021-00082-y.