Caloric Restriction Slows Ovarian Aging in Monkeys
In Aging, researchers have published their discovery that three years of caloric restriction in rhesus macaques that are beginning to enter menopause slows their ovarian aging.
Closer to people than mice
The ovary is one of the first human organs to be seriously affected by aging, with the follicles becoming depleted long before most causes of age-related mortality become common [1]. However, the associated loss of estrogen has downstream consequences on many other organs [2].
There are crucial caveats to studying ovarian health in mice, as their estrous cycle is different from that of women; mice do not menstruate. Rhesus macaques, on the other hand, are much closer to people; they live much longer, share 93% of our genome, and have a menstrual cycle that, while having a seasonal component [3], is broadly similar to that of women. Previous work has found that caloric restriction improves the healthspan and lifespan of these monkeys [4], but that work did not investigate its effects on ovarian health.
Benefits just before menopause
These researchers tested the effects of three years of 30% caloric restriction on female rhesus macaques. The young group began the study at 7-10 years old (a few years after puberty), and the old group began the study at ages 16-23. All of the young animals began this study with regular menstrual cycles, while only one old monkey in the control group and none in the caloric restriction group had regular cycles. This was not a lifespan or healthspan study; the monkeys’ organs were harvested after completion.
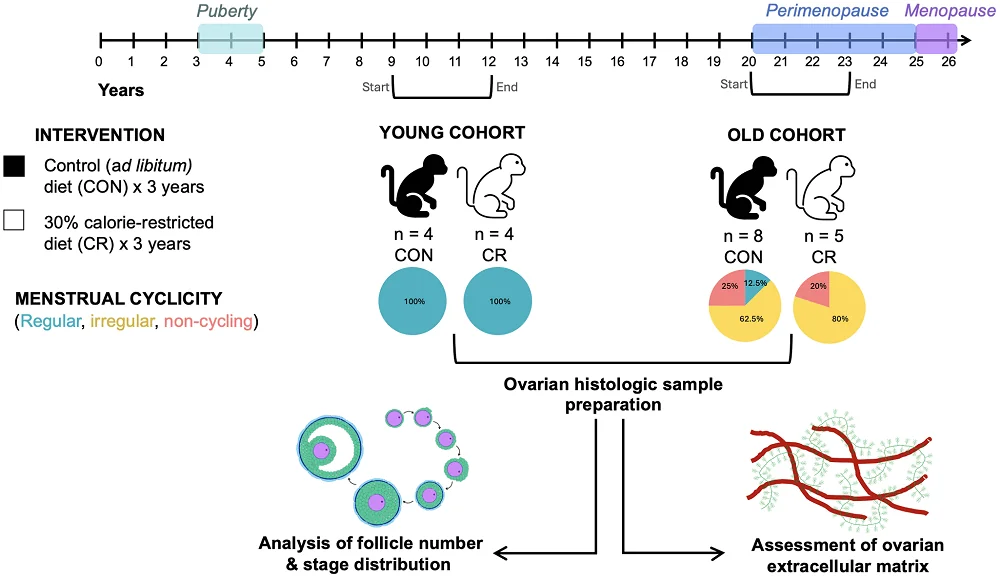
The comparisons between young and old animals were entirely as expected: older animals had significantly fewer follicles of every type than their younger counterparts, and menopause visibly shrank the ovaries. Unsurprisingly, there was an extremely strong correlation between follicle count and menstruation status.
In irregularly cycling (perimenopausal) animals, the number of quiescent (primordial) and multilayer follicles was greater in monkeys on caloric restriction than in the control group, with positive trends in many other follicle groups, leading to an increase in overall follicle density and suggesting a general increase in ovarian activity. Additionally, the proportion of follicle classes in the caloric restriction group was more like that of young animals. However, no benefit to follicle density was found in menopausal monkeys.
The ovaries normally become more fibrotic with age, which was found to occur in the control group but not in the caloric restriction group, as measured by tissue staining. Similarly, there is a loss of hyaluronic acid with age, which was only significant in the control group. These results suggest that caloric restriction delays age-related ovarian fibrosis.
A small study that confirms existing knowledge
This was a relatively small study, and the researchers note that their three years of caloric restriction are, in the research world, considered short term, as other studies on lifespan and healthspan have involved lifelong dietary restrictions. They acknowledge that it is difficult to impossible for human beings to stay on such a restrictive diet, and they suggest that such techniques as intermittent fasting may be more practical and that caloric restriction mimetic drugs may be of use, although further study is required in both of these areas.
Furthermore, most of this study’s results were based on computer analysis of histological slides; there was no investigation into the biochemistry behind the effects of caloric restriction on ovarian function. The researchers hypothesize that the effects are similar between people, monkeys, and mice, such as the sirtuin pathway [5].
The researchers hold that the timing of caloric restriction may be crucial. The significant effects were seen in monkeys whose ovaries were beginning to experience age-related decline. Caloric restriction to this degree did not restore the function of fully menopausal monkeys, and it did not seem to have any significant effects on the ovaries of younger monkeys; it is likely that these results are also true for humans. Therefore, for comparable interventions to be appropriate for the maintenance of ovarian health, they would need to be administered at a very specific time in a woman’s life.
Literature
[1] Hansen, K. R., Knowlton, N. S., Thyer, A. C., Charleston, J. S., Soules, M. R., & Klein, N. A. (2008). A new model of reproductive aging: the decline in ovarian non-growing follicle number from birth to menopause. Human reproduction, 23(3), 699-708.
[2] Cui, J., Shen, Y., & Li, R. (2013). Estrogen synthesis and signaling pathways during aging: from periphery to brain. Trends in molecular medicine, 19(3), 197-209.
[3] Beltran-Frutos, E., Casarini, L., Santi, D., & Brigante, G. (2022). Seasonal reproduction and gonadal function: A focus on humans starting from animal studies. Biology of Reproduction, 106(1), 47-57.
[4] Mattison, J. A., Colman, R. J., Beasley, T. M., Allison, D. B., Kemnitz, J. W., Roth, G. S., … & Anderson, R. M. (2017). Caloric restriction improves health and survival of rhesus monkeys. Nature communications, 8(1), 14063.
[5] Long, G. Y., Yang, J. Y., Xu, J. J., Ni, Y. H., Zhou, X. L., Ma, J. Y., … & Luo, L. L. (2019). SIRT1 knock-in mice preserve ovarian reserve resembling caloric restriction. Gene, 686, 194-202.







 : ECM drives tissue regeneration and wound healing in all human organs. In acute or chronic organ injuries, especially in the aging population, abnormal ECM healing leads to fibrosis, inflammation & ischemia. XM Therapeutics’ platform technology provides a micro-molded non-adhesive substrate to culture cells in 3D, which allows human ECM particles produced in vitro to repair the abnormal ECM. It has shown that ECM induces recovery of ischemic myocardium by upregulating the protein networks involved in cellular contractility and metabolism.
: ECM drives tissue regeneration and wound healing in all human organs. In acute or chronic organ injuries, especially in the aging population, abnormal ECM healing leads to fibrosis, inflammation & ischemia. XM Therapeutics’ platform technology provides a micro-molded non-adhesive substrate to culture cells in 3D, which allows human ECM particles produced in vitro to repair the abnormal ECM. It has shown that ECM induces recovery of ischemic myocardium by upregulating the protein networks involved in cellular contractility and metabolism.