Like many people, I’m both wary of and intrigued by people who make bombastic claims. Years in the longevity field have taught me caution but also that big claims are not necessarily outlandish, and few people make bigger claims than Joshua “Scotch” McClure, founder and CEO of Maxwell Biosciences. The company developed Claromers™, synthetic small molecules that mimic the natural immune system. According to McClure, his company’s synthetic version of the ubiquitous but naturally unstable anti-microbial peptide LL-37 can fend off almost any infection, potentially leading to a considerable increase in human healthspan and lifespan.
McClure is a fixture at geroscience conferences, where he pushes his company’s simple yet revolutionary idea with typical fervor, and he has some serious proof to back it up. He raises eyebrows, but he also raises money. Maxwell has entered numerous collaborations with the US military and countries like India. It also recently conducted successful non-human primate trials, a perfect prelude to the upcoming clinical trials. All of this meant one thing: it was time for us to have a chat.
People in our field who are not trained biologists usually have the most interesting stories of how they got here. What’s yours?
Ever since I was a kid, I’ve been very interested in genetics. I’ve been reading genetics books and scientific publications for a long time. I became very interested in longevity science when the Conboys started publishing about the benefits of heterochronic parabiosis. This convinced me that there was something in the plasma that would allow us to live longer.
I was working in commercial real estate AI at the time, making a lot of money. I thought this might be something I could fund as a lab or a side project, but then my dad and my daughter got really sick with antibiotic-resistant infections. One was a viral infection; the other was a bacterial infection.
At that point, I couldn’t look at healthspan as a side project anymore. This was a major emergency in my family. I had to take a leave of absence from my job to figure out what was going on with my daughter and my dad. Eventually, they both pulled through.
I understand that you’re a scientist, just not a biologist.
Yes, I’m a data scientist. Not a biologist, though I was always into physics and genetics. You could call me a biophysicist – understanding electromagnetic fields and electrostatic interactions of biology. That’s the kind of stuff I was studying on the side.
I looked at various business models, and the one I ultimately selected was mass-scale, hyper-affordable medicine. If you’re going to raise the average lifespan of humanity, the way to do it is by making just about everybody immune to whatever the biggest killer is. And the biggest killer is infectious disease, by far.
Pretty much everybody has chronic subacute infections, such as Epstein-Barr virus. Subacute means it hasn’t popped up above the radar. You’re not feeling sick, but you don’t know what your highest level of performance is. You wake up in the morning, you’re a bit groggy, so you take some coffee, and then you feel better. That could very well be Epstein-Barr virus, or herpes, or whatever.
You need a certain amount of energy to function. Underneath all the active operations are passive operations – your heart beating, your breathing, your thought processes – but also maintaining homeostasis, which draws core energy. This is like a spaceship’s life support system. If life support fails, the body dies, because it’s constantly being attacked by bacteria.
E. coli, Staphylococcus live inside us. They are our enemies; they just have their swords sheathed. They’re ready to eat us just the same way they’re ready to eat a hamburger coming through our gastrointestinal system, because we are essentially a hamburger.
The reason they’re not hurting us is because of our innate immune system peptides that reside inside the mucosal membranes of the gastrointestinal system, the nose, eyes, everywhere. They’re like pokey swords that are holding the bacteria and all the microbiome at sword point. If they go pathogenic, there’s an immediate attack within less than a second, so they don’t.
But as soon as you get weaker because of stress or old age, and you’re not able to have quite so many swords out there poking at them, they think, “He’s not really paying attention.” And then suddenly you get ulcerative colitis, gut permeability, and that’s the slow chronic disease decline that we’re trying to get rid of.
That may be one of the sources of inflammaging.
Correct. That’s the fundamental, multi-million-year competition between the tiny and the big, the ongoing battle that goes far beyond our species. If you look at sheer biomass on the planet, humans and all mammals don’t even show up on a graph. About 98 percent of the planet’s biomass is fungi and bacteria. It’s not a safe place for us to live.
The major thing killing us is infectious disease. It’s life eating life. And we are creating a synthetic immune system that is much better than your innate immune peptides and makes it impossible for you to be eaten when you get stressed. You still have all those blades out there keeping your E. coli in check, and you also completely get rid of the toxic microbiome. That’s where I think we’re headed – getting rid of infectious disease.
So, you have a technology that can mimic peptides and you’re specifically interested in the antimicrobial peptide LL-37. I understand it took you a long time to sift through a huge peptide database and zero in on LL-37 as a major regulator of the immune system.
Yes, and when we figured that out, we thought we had discovered something really amazing, but then we found that LL-37 is one of the most studied peptides in the human body. There are thousands of published articles about it.
So, I only discovered something for myself. Many bioengineers, chemists, and biologists knew that LL-37 is important, but people in our field also tend to think infectious disease doesn’t matter. There are sexier things like mapping the genome. People don’t care about some peptide that everybody has.
But the abundance of previous knowledge corroborated your science.
Yes, it totally corroborated it. Then I got involved with some of the top minds in what are called defensins, the antimicrobial peptides. I started talking with them, and they told me about this invention where the Department of Energy and DARPA got together to mimic peptides with a small molecule form factor.
How does this peptide actually work against pathogens?
It’s a positively charged linear peptide that looks like a corkscrew. It’s attracted to negatively charged pathogens, which includes almost all of them. Then, this positively charged corkscrew just drills into the pathogen and rips it apart. The only way a pathogen has a chance is to keep the peptide away or avoid detection.
LL-37 works against both Gram-positive and Gram-negative bacteria and enveloped viruses, and humans only have this one core peptide for their innate immune system. If it didn’t work against everything, you’d be dead. A lot of people don’t produce enough of it – babies, senior citizens, people not getting enough exercise, sunlight, good nutrition – so they are vulnerable.
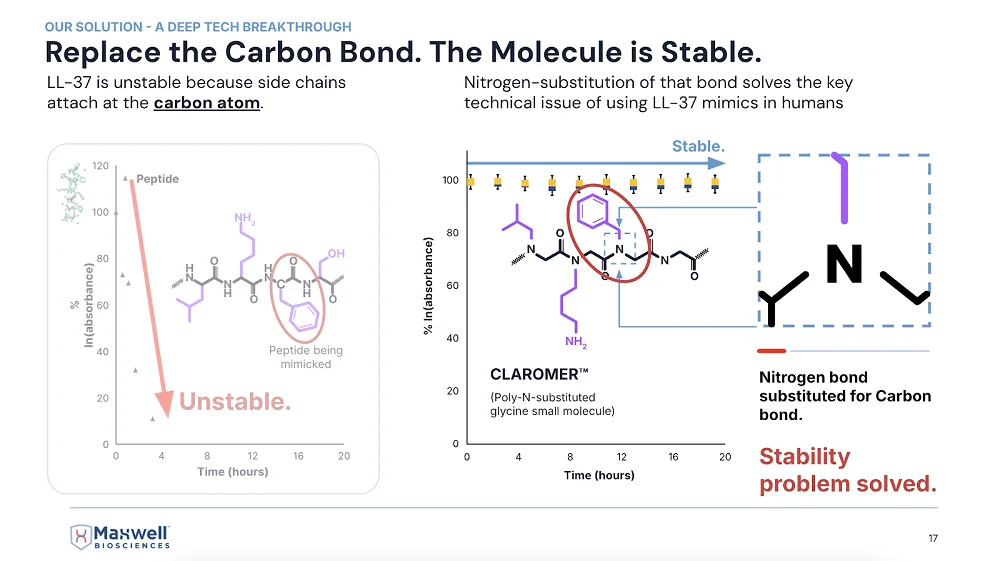
I understand that LL-37 is as unstable as it is important, which was the main problem you had to overcome.
Yes, it’s like wet toilet paper – it falls apart very quickly.
So, you realized that to use it, you must stabilize it somehow, and you went for the peptoid architecture?
That’s right. We took the functional side chains that do all the work on the peptide and did a lot of extra math. We went through many very frustrating years of difficult work to do something that you’d think would be pretty easy: attaching the side chain to the nitrogen on the peptide’s backbone instead of the alpha-carbon.
The math of replicating the biological function of a peptide – that’s the thing. You wonder how that peptide works, and then you find out that even with thousands of published articles on it, people don’t really know that.
A slide from McClure’s presentation at last year’s ARDD conference in Copenhagen
Are Claromers™ your proprietary version of peptoids?
Yes, the antimicrobial version of peptoids. We’ve got lots of new ones coming out that are way better than the ones we’re taking through the FDA right now, but even those are so incredible that the FDA loves them.
First, there is no toxic concentration that we have found yet. Second, they’re anti-inflammatory and extremely broad spectrum. We have one that works against bacteria and fungi, and another against viruses: all influenza, all coronaviruses, SARS-1, SARS-2, regular cold. It’s literally the cure for the common cold. It’s crazy but amazing.
What about long COVID?
It’s going to knock it out completely. I have no doubt about it. Of course, these are my opinions. I’m the CEO of the company, and I am paid to be the optimist. But these are not promissory statements. And I’m not asking for investment. We don’t need it.
The fact is that, in partnership with the government, we’ve passed non-human primate studies with a single molecule killing the worst type of bacteria and the worst fungus we could select – the worst multi-drug-resistant versions of both that would kill you because there’s no drug to treat it.
You barely need diagnostics anymore. Just use this all the time, and if it doesn’t work, then you can do some expensive diagnostics to figure out what wild thing is in there that isn’t being killed. It kills all the bad things and leaves the good stuff in the microbiome.
Because of how it mimics the natural thing?
Yes, it’s exactly the same natural mechanism of action, so it leaves your microbiome alone.
You’ve already touched on this, but let’s talk some more about how this relates to aging.
Infectious disease is the primary driver of aging. It is the primary disruptor of homeostasis. As soon as we start fighting infectious disease with our drug – the first time we’re approved by the FDA for any kind of infectious disease – it will start lengthening healthspan immediately.
I think it’s inevitable at this point because we passed the non-human primate study. The only thing that could stop us now would be lack of cash flow, but we don’t have a problem with that. We’re working on multiple big contracts right now with the US military, the governments of India, UAE – direct government contracts. We’re working on two big contracts with commercial companies.
This is going to be world-shaking. Just like AI – even though it’s not currently replacing your job, you can pretty much bet it’s going to replace most jobs. Same with this technology: it’s going to eradicate infectious disease and more.
The first thing that came to my mind when I heard about your technology is that infections are an insanely prevalent cause of death in the oldest old. Many think this is a major factor that limits our maximal lifespan.
It truly is. Many times, people say, “Great-grandma fell and broke her hip,” and everyone thinks that’s natural, that’s how very old people die, but why does breaking a leg or a hip make someone fall apart and die?
It’s because their body has been fending off infectious disease pretty well up to this point. Now it must redirect just a little bit of energy to healing the hip, and not enough is left to defend against infectious disease. You’re breathing millions of fungal spores every day that will eat you just like a rotten tree if you don’t produce enough of this peptide to keep them destroyed.
This big change – pretty much eliminating the ever-present attack of pathogens against us – is going to greatly expand our healthspan. I’m predicting 20 to 40 additional years for the whole population just from our drug, comparable to what advances against infectious diseases have already done for lifespan.
Once we solve this, once we clean everything up and you’re like, “Okay, bird flu is a big issue right now, but I don’t need to worry about it because I’ve got this drug; everything is clean – my food is clean, my water is clean, my air is clean,” – then you can start sort of moving up Maslow’s hierarchy of needs and getting into the sexy stuff – can I enhance my vision? Can I enhance other things?
How does a collaboration with a country like India work?
Countries have their own priorities and budgets established for those priorities. With India, it’s for rare, neglected tropical diseases. This budget has been around since 2017, but no one has gotten it because no one wanted to tackle the indications they want us to address.
With India, it’s more about antibiotic-resistant septic infections that happen out there in the villages. India has plenty of antibiotics – they’re the home of generic antibiotics. What they need is help with difficult situations where somebody is going to die before they can get diagnostics. There’s no diagnostics out there to tell you what type of bacteria it is, so you must have something very broad-spectrum at hand to cure whatever it is.
The whole pharma industry says, “No, that’s not how medicine works.” We come in and say, “This is exactly how medicine should work.” Our product is shelf-stable and thermal-stable. It can be hot enough to cook an egg – and our drug will still be effective.
It’s a $300 million deal over three years, and it’s helping us while we pursue bigger things like tuberculosis and long COVID. It’s non-dilutive funding, essentially.
You saw no toxicity in your preclinical studies?
Zero toxicity. Not only it’s not toxic to human cells, but it’s not toxic to the commensal microbiome. We specifically measured the commensal microbiome in rhesus macaque monkeys in the India project, where we went up against methicillin-resistant Staphylococcus aureus (MRSA).
In the microbiome test, we found that they have a huge amount of Staphylococcus aureus in their sinus cavity – it’s one of the predominant bacteria there. Our compound works great against Staph aureus, but all our data said it doesn’t affect the commensal microbiome.
So, we asked: does that mean it’s selective for commensal Staph aureus and only kills the pathogenic Staph aureus? That seemed crazy, but that’s what we found. The commensal Staph aureus was untouched and in fact helped to protect the primate against the infection.
We had to remove some of the commensal microbiome to even give our MRSA strain a seat on the bus, so to speak. It took us a long time to actually establish an infection. This tells me that many human infections have to do with microbiome disruption. We eat preservatives and other things that disrupt the protective microbiome, which is a major way to get infected. The good bacteria form a shield that prevents pathogenic bacteria from getting through.
There must be some mechanism that prevents the peptide or peptoid from attacking beneficial bacteria. Do you know what it is?
We do, and we’ve published on it. Like I said, LL-37 is extremely well studied. The mechanism of action of LL-37 and our compounds is that they target negatively charged phospholipids on the membrane of bacteria, fungi, or viruses. That negatively charged phospholipid in the membrane is required to merge with a human cell by connecting to TIM receptors (TIM-2, TIM-3, TIM-4). So, it’s a hallmark of aggressive pathogens.
The negatively charged phospholipids are present in the membrane of the commensal microbiome, too, but they are covered with a V-shaped or shield-shaped molecule called annexin-5 (and the whole annexin family). The annexin goes over the top of the negatively charged phospholipids, saying “I’m not going to attack you.”
If they take their annexin-5 off, they become pathogenic. That’s how Staphylococcus stays commensal as long as it’s getting fed inside your gut. But if you stop eating and stop producing antimicrobial peptides, it’s like “You’re what’s for dinner” – it comes after you.
Anything you can tell me about your collaboration with the military?
They want things like prophylactic wound care for combatants on the run; it’s pre-first aid, where you can squirt something into a puncture wound to stop bleeding and potential infection.
We have about seven Collaborative Research and Development Agreements (CRADAs) with the Pentagon. Some are for quite exotic things like anthrax and Ebola, things that could be weaponized. The military says, “We have to have something against that, and we’re willing to pay a billion dollars to stockpile it, just in case somebody uses a weaponized version against us.”
We’re also working on more common things, such as anti-diarrheal applications, which we could use for commercial purposes as well. How many battles have been won by dysentery rather than by the enemy army?
How far out are you in terms of human trials and approvals?
This year, we got the data back on the non-human primate studies, which is pretty much equivalent to a human Phase I. It was totally safe and very effective, so we have a very high probability for the human trials outcome. The value of the company has gone up significantly, and the certainty of the outcome has increased, which is why we’ve gotten all these contracts with governments and companies.
We would like to ask you a small favor. We are a non-profit foundation, and unlike some other organizations, we have no shareholders and no products to sell you. All our news and educational content is free for everyone to read, but it does mean that we rely on the help of people like you. Every contribution, no matter if it’s big or small, supports independent journalism and sustains our future.