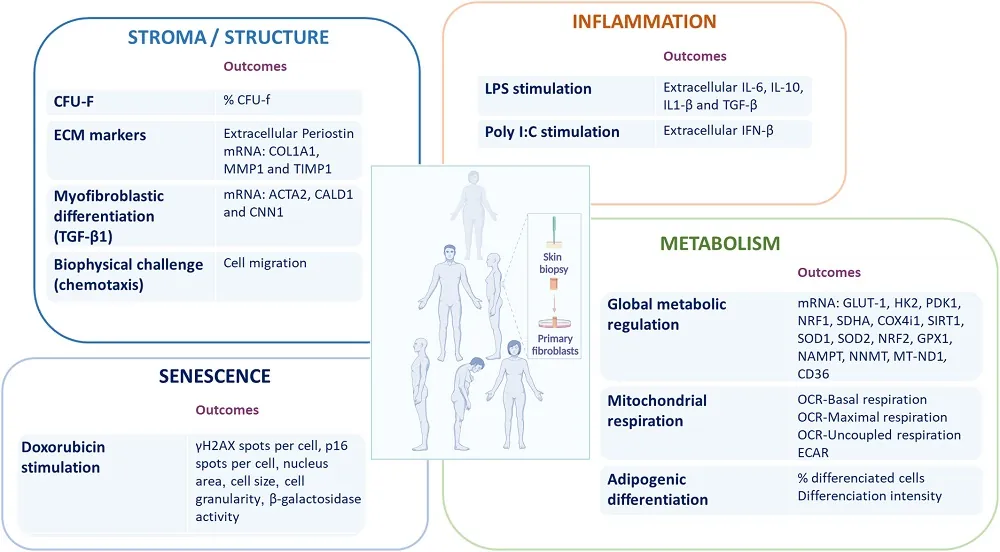
Scientists Successfully Edit Mitochondrial DNA
A new study demonstrates that novel gene-editing tools can correct disease-causing mutations in mitochondrial DNA in primary human cells [1].
Smaller editing tools needed
Genome-editing tools such as CRISPR were one of the greatest scientific breakthroughs of this century. However, they are only good for editing nuclear DNA.
Mitochondria, the energy-producing organelles, have their own circular DNA (mtDNA) that resides inside each mitochondrion and codes for a number of essential proteins. Mutations in mtDNA cause several diseases and are also associated with aging [2]. Until very recently, there was no easy way to edit mtDNA since CRISPR-based tools are too large to enter mitochondria.
The situation began to change with the introduction of smaller editing tools, but more research is needed to test and refine them. In a new study published in PLOS Biology, scientists from the University Medical Center Utrecht in the Netherlands used the double-stranded DNA deaminase toxin A-derived cytosine base editor (DdCBE), paired with guiding proteins called TALE, “to develop in vitro disease models and assess therapeutic strategies for mitochondrial diseases in primary human cells.”
Creating a disease model
First, the team used DdCBE to introduce a loss-of-function mutation (m.15150G>A) in human primary adult liver stem cell-derived organoids. This particular mutation has not been associated yet with any known disease, but other mutations in the same gene (MT-CYB) have. The researchers report that their editing tool successfully introduced the mutation.
This is an important step in creating models of mitochondrial diseases so that they can be studied and cured. “Mitochondrial dysfunction and mtDNA alterations are implicated in several age-associated pathologies, however, our ability to understand the underlying mechanisms is limited by lack of appropriate models,” said Dr. Amutha Boominathan, a senior researcher at the Lifespan Research Institute, who was not involved in this study.
While a cell has only two copies of the nuclear DNA, one from each parent, there can be hundreds of thousands of mitochondria in each cell, each one with its own circular DNA. Therefore, an edit needs to be introduced to as many of those copies as possible. The presence of more than one type of mtDNA within a single cell is called heteroplasmy.
When the researchers introduced a pathogenic mutation into healthy liver organoids, they did not create cells that were 100% mutated. Instead, by isolating and growing single cells, they generated a collection of organoid lines with a wide range of heteroplasmy levels (from 0% to 80% mutated). This allowed them to study the effects of different levels on the severity of the disease, as naturally occurring DNA diseases also manifest themselves only past a certain heteroplasmy threshold.
Mutation fixed
The next step was to try fixing a known harmful mutation. In fibroblasts from a patient, the DdCBE system successfully corrected the pathogenic m.4291T>C mutation, which is linked to Gitelman-like syndrome, a group of inherited kidney disorders.
Heteroplasmy remained a challenge: when the researchers grew out colonies from single edited cells, they found a wide range of DNA correction levels. On the bright side, those levels remained stable over 50 days of follow-up and even slightly increased, showing that the corrected mitochondria were healthy and not at a selective disadvantage within the cell.
In cell lines with a high level of correction (76% and 81%), the mitochondrial membrane potential was successfully restored to the level of healthy control cells, suggesting functional rescue. In a line with low correction (35%), there was no improvement.
The results for overall energy production were more modest and inconsistent. While slight improvements were observed in some experiments, the effect was not as strong or reliable as the restoration of the membrane potential. The authors note that this warrants further study.
Initially, the team used a tried-and-true method of delivery: DNA carried by viral vectors. In later experiments, they showed that a better method was to deliver the editor as modified RNA (modRNA). The modifications included tweaking RNA nucleotides for greater stability and shielding the molecule from being detected by the immune system. Compared to DNA delivery, modRNA demonstrated much higher efficiency and less cytotoxicity.
The modified RNA molecules were delivered using lipid nanoparticles (LNPs). This is the same state-of-the-art technology used to deliver the mRNA in COVID-19, considered the most advanced non-viral system for in vivo delivery.
“Adapting precision DNA editing tools such as base editors to target the mitochondrial genome holds significant promise for both modeling and treating mitochondrial DNA (mtDNA) mutation-associated diseases,” said Dr. Boominathan. “However, this approach faces several challenges, including the high number of edits required per cell (due to the large mtDNA copy number), achieving homogeneous editing across cell populations, and minimizing off-target effects. In this study, the authors successfully generated a pathogenic mutation in liver organoids and corrected the m.4291T>C mutation in patient-derived fibroblasts. Nonetheless, limitations such as variability in editing efficiency – both in the extent and uniformity of edits – persist and warrant further optimization.”
Literature
[1] Joore, I. P., Shehata, S., Muffels, I., Castro-Alpízar, J., Jiménez-Curiel, E., Nagyova, E., … & Koppens, M. A. (2025). Correction of pathogenic mitochondrial DNA in patient-derived disease models using mitochondrial base editors. PLoS biology, 23(6), e3003207.
[2] Sprason, C., Tucker, T., & Clancy, D. (2024). MtDNA deletions and aging. Frontiers in Aging, 5, 1359638.