Since 2009, the support of curated aging research projects has been a major part of Lifespan Research Institute’s commitment to changing the way the world researches and treats age-related disease.
This project builds upon the earlier work of the Boominathan Lab at Lifespan Research Institute.


Principal Investigator: Amutha Boominathan
As we age, the immune system’s ability to recognize and eliminate senescent cells deteriorates, allowing these pro-inflammatory cells to persist and drive chronic diseases.
Despite their limited numbers, senescent cells exert disproportionate influence over tissue health, regenerative capacity, and immune competence. Intervening at this node—by clearing senescent cells, blocking their harmful secretions, or restoring immune surveillance—offers a uniquely powerful lever to reverse or prevent multiple age-associated pathologies at once.
Efforts to identify senolytics – drugs that selectively eliminate senescent cells, have so far yielded candidates with limited translational potential, most likely due to limited efficiency in senolytic activity. Therefore, optimizing the efficiency of senolytic interventions is a key step towards determining the extent of the contribution of cellular senescence towards aging and in developing classes of drugs that would be effective in treating senescence-associated diseases of aging.
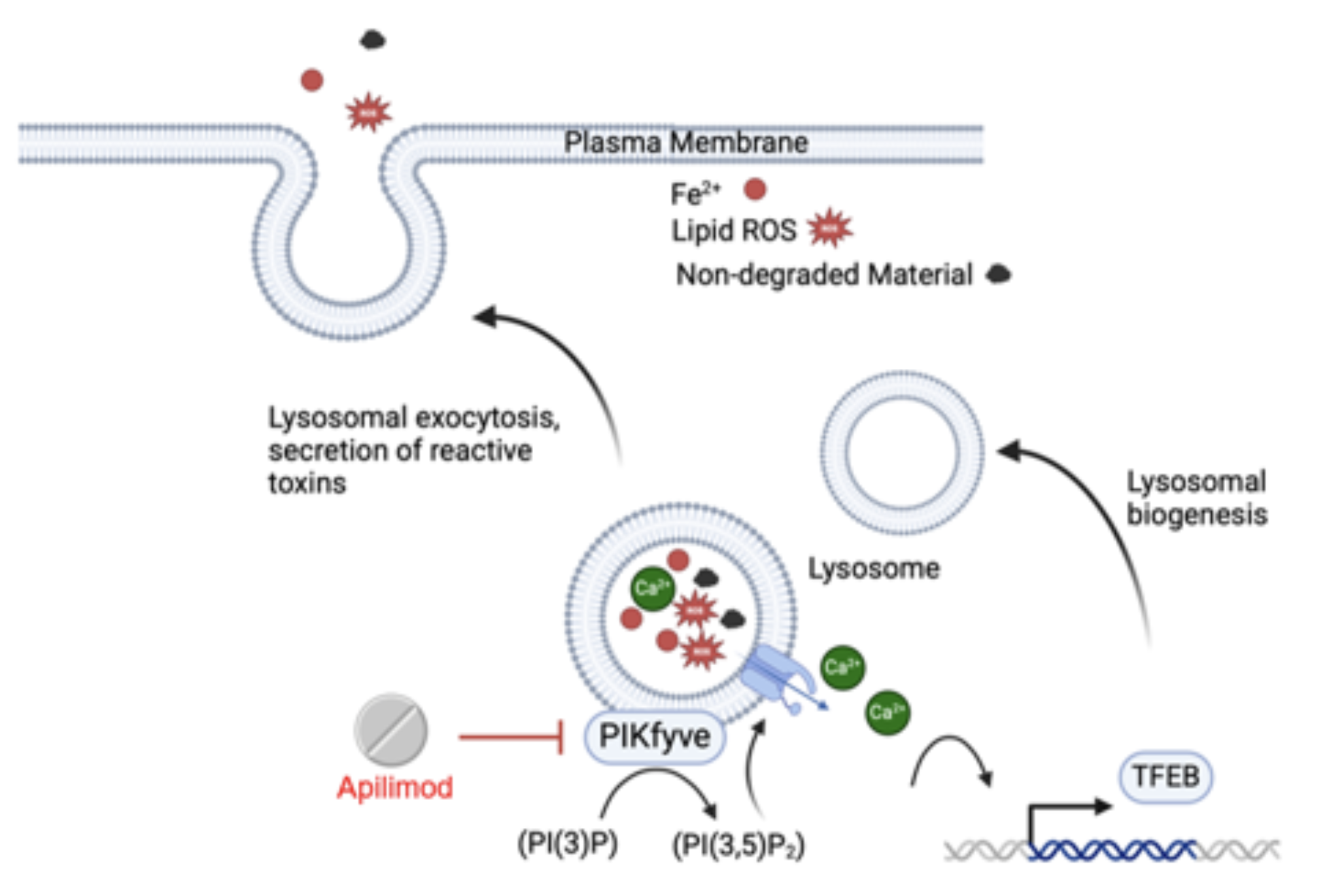
Recently we characterized the senescent cell surface proteomic landscape and identified proteins that are preferentially expressed on the plasma membrane of senescent cells. Many of these proteins are lysosomal enzymes, pointing to lysosomal exocytosis as a likely mechanism that leads to their persistent display on the cell surface.
Our research has further determined that blocking lysosomal exocytosis via PIKfyve kinase inhibition with a repurposed small molecule drug, apilimod, results in selective killing of senescent cells in vitro, while not affecting quiescent and proliferating cells (see figure). Apilimod has also been safely administered in vivo and effectively removes senescent cells and reduces tissue remodeling in a bleomycin mouse model of pulmonary fibrosis. We are presently designing experiments to further test additional apilimod based analogues that have improved pharmacokinetic profiles in anticipation of near-future clinical trials.
This project builds upon the earlier work of the Sharma Lab at Lifespan Research Institute.

Principal Investigator: Amit Sharma
Principal Investigator: Amutha Boominathan
As we age, the immune system’s ability to recognize and eliminate senescent cells deteriorates, allowing these pro-inflammatory cells to persist and drive chronic diseases. We are investigating how aging disrupts immune surveillance and how it can be restored to promote healthy longevity.
Senescent cells secrete cytokines and ligands that normally recruit immune cells, particularly natural killer (NK) cells, to eliminate them. However, aging impairs this clearance mechanism. Aged NK cells exhibit reduced cytotoxicity, altered receptor expression, and diminished ability to respond to senescent ligands, allowing senescent cells to evade immune attack and accumulate in tissues. This immunosenescence contributes to the development of persistent inflammation, fibrosis, and impaired tissue repair.
We’ve developed ex vivo platforms to enhance human NK cell activity against senescent targets, demonstrating that NK function can be rejuvenated and leveraged therapeutically. We also identified γδ T cells as novel senolytic immune effectors that recognize senescent cells through stress ligands such as BTN3A1 and NKG2D, functioning independently of MHC. These findings offer powerful opportunities to engineer cell-based immunotherapies that can remove senescent cells, even in aged or immunocompromised hosts.
In parallel, we explore how senescent cells blunt inflammation resolution. This bidirectional crosstalk between senescent cells and immune cells represents a critical node in age-related tissue dysfunction.
We discovered that human γδ T cells (Vγ9Vδ2) selectively eliminate senescent cells through recognition of BTN3A1 and other ligands. Adoptive transfer of γδ T cells into IPF models reduces senescence burden and fibrosis, offering a cell-based senolytic strategy. [bioRxiv, 2025]
We developed a robust, physiologically relevant NK co-culture platform to study senescence immunosurveillance. Enriched human NK cells exhibit enhanced and selective killing of senescent cells, facilitating drug screening and the development of immunotherapy. [Aging, 2022]
By decoding these interactions, we aim to rebuild immune-mediated senescence surveillance, enhance tissue homeostasis, and develop the next generation of immunotherapeutic strategies against aging.
Principal Investigator: Amit Sharma
The aging brain is vulnerable to invasion by infectious microbes, and there is compelling evidence that some of these pathogens drive neurodegenerative aging of the Alzheimer’s type (AD). One of the ways these microbes promote the disease is by provoking brain neurons to release beta-amyloid, which appears to act as an antimicrobial peptide (AMP). That might be fine if the brain’s resident immune cells (microglia) followed up by clearing out the amyloid-microbe complex. But over time, the burden of pathogens and beta-amyloid aggregates overcomes the microglia, and beta-amyloid accumulates and begins driving neurodegenerative changes. Meanwhile, as the demands on the microglia progressively outstrip their abilities, they become increasingly aggressive and unruly, ultimately setting the brain ablaze with inflammation and destroying the very neurons that they exist to protect.
Research Highlights:
Lifespan Research Institute is sponsoring work by Dr. Annelise Barron to develop dual-action drugs that could simultaneously tackle the two key forms of damage in this scenario: the microbes that infiltrate the brain and the beta-amyloid that is in part released to disable them. Much of Dr. Barron’s biomedical engineering research is on peptoids — modified versions of short protein-like structures that allow them to persist longer in the body and modify their functions. For this project, she is working on peptoids that will mimic the natural AMP LL-37, which is produced in humans and helps clear pathogens through a variety of mechanisms.
The first benefit of the optimized peptoid would be to destroy key pathogens implicated in AD, thereby preventing them from damaging the brain and making the release of beta-amyloid to contain the infection unnecessary, which would in turn lower the brain’s exposure to the aggregates.
The optimized peptoid would also safely mimic a a second property of LL-37 identified by Dr. Barron and Dr. Jevgenij Raskatov: the ability to safely complement and ‘tame’ beta-amyloid. LL-37 seems in some ways to be a pairmate to beta-amyloid: the two molecules’ molecular weights are almost identical to one another’s, and their specific amino acid sequences and opposing charges create strong and selective binding interactions. When the two are in each other’s presence, LL-37 and beta-amyloid strongly bind to each other and hold each other safely in check, thereby blocking one another’s toxicity. It may not be a coincidence that LL-37 levels in the brain decline with age, roughly coinciding with the rise in beta-amyloid levels that occurs up until the verge of clinical dementia.


Principal Investigator: Annelise E. Barron
Research Team: Kristian Sørensen, Jennifer Lin, Josefine Eilsø Nielsen, and John Fortkort
Our large arteries are exquisitely engineered with concentric layers of elastic mesh comprised of the protein elastin that give them life-sustaining mechanical properties. First, the elastin web protects our brains and kidneys by cushioning the rhythmic hydraulic pounding of blood surging out of the heart. Second, it acts as a “pressure reservoir” that sustains blood flow to those same organs and others when the heart is between beats.
But arterial elastin is continuously mechanically damaged by the relentless cycles of stretch and relaxation with each heartbeat, and this mechanical damage is exacerbated by the action of enzymes and free radicals that damage and degrade it. As the elastin structure frays, it loses its cushioning capacity, stiffening our arteries and exposing our kidneys and brains to the destructive hammering of the pulse while leaving us vulnerable to aneurysms, strokes, and vascular dementia.
Unfortunately, our bodies did not evolve systems to maintain our arterial elastin structures in the exquisite order of our youth throughout the many decades that modern humans live, let alone to enable much longer and healthier ones. We have no effective means to repair frayed elastin structures, and the cells that spin the elastin mesh during early development stop doing so by the time we are adults. And drugs that researchers have reported to activate elastin synthesis either do not do so in the artery or stop doing so early in our development.
Research Highlights:
To regenerate the aging artery, LRI is funding an innovative strategy from Dr. Ziyu Wang at the University of Sydney. USYD researchers discovered that they could stimulate skin cells from old donors to start synthesizing mature elastin again by simply supplying them with bioengineered elastin precursors. With this semifinished material, Dr. Suzanne Mithieux (an advisor of Dr. Wang in Dr. Weiss’ lab) showed that old skin cells can create an elastin layer appropriate to new skin on the interface of surgical skin repair patches.
To see if this trick could similarly stimulate cells in the arteries to spin out the more complex elastin structures in the middle layer of the arterial wall, Dr. Wang implanted elastin precursors enmeshed in a biodegradable polymer into the arteries of mice. With the elastin precursors at hand, cells inside the mice’s vessels began to produce new arterial elastin with the appropriate structure, properly aligned with the artery’s smooth muscle cells. A cell culture study using aortic smooth muscle cells supported that these cells could contribute to the process only if given elastin precursor.
To noninvasively deliver this critical material into aging arteries, Dr. Wang is encapsulating it within regenerative nanostructures that are coated with antibodies that bind to damaged elastin scaffolding. Once they hone to the damaged sites, the particles are expected to release the elastin precursor locally, enabling nearby cells to begin producing new elastin again, which Dr. Wang’s pilot experiment suggests will regenerate the artery’s life-preserving elastin structures.
Principal Investigator: Ziyu Wang
Research Team: Anthony Weiss (Co-PI), Dr. Suzanne Mithieux, Kekini Kuppan

Of all the challenges in cell therapy, replacement of neurons in the neocortex is both the most important (the brain being the seat of consciousness and identity) and perhaps the most formidable. Only recently have any researchers succeeded in integrating new neurons into this area of the brain. Moreover, the vast majority of transplanted cells in these cases have failed to survive, and the few survivors have achieved only limited function and integration into existing circuits.
Research Highlights:
LRI has sponsored Dr. Jean Hébert’s work to advance an innovative strategy to address this challenge. Dr. Hébert’s team has transplanted neuronal precursors along with precursors of the blood vessels needed to nourish and enhance the survival and integration of new neurons. Initial analysis shows that this approach is successful.
However, as transplanting neurons throughout the entire aging neocortex would be extremely invasive, posing serious risk of injury to a tissue we cannot afford to damage, the team has engineered microglia (which, unlike neurons and their precursors, are highly mobile cells) to disperse widely from the site of transplant. These microglia can then be reprogrammed into cortical projection neurons at their destination.
Initial work on the transdifferentiation of microglia into neurons has been established. From there, the team will characterize the integration of the transplanted microglia-turned-neurons into host circuits of converted neurons derived from our engineered transplanted microglia. It has been demonstrated that depletion of microglia can enhance the process and facilitate good dispersion of newly transplanted microglia; Dr. Hébert’s team aims to determine whether depleting host microglia enhances these processes in different models.

Principal Investigator: Jean Hébert, PhD
Research Team: Hiroko Nobuta, Joanna Krzyspiak, Alexandra Quezada, Marta Gronska-Peski, Jayleecia Smith
Prof. Grune is the Scientific Director of the German Institute of Human Nutrition and has been working on protein degradation of damaged proteins and aging.
Lipofuscin (LF) is a strongly oxidized material composed of covalently cross-linked proteins, lipids, and carbohydrates. Cellular LF increases with age and negatively correlates with the remaining life span of cells. Lipofuscin accumulation is especially pronounced in postmitotic cells (including cardiomyocytes and neurons) as these cells are unable to “dilute” their lipofuscin via cell division. LF by itself impairs cardiomyocyte function by declining its contractility. Importantly, no known mammalian enzyme degrades lipofuscin, therefore LF accumulates within the cell, mostly within the lysosomes.
Microorganisms, particularly bacteria, possess a wide array of enzymes that allow the degradation of any conceivable molecule formed in nature. The project, therefore, aims at identifying bacterial enzymes able to degrade LF. The project includes the following tasks:
Research Highlights:
Prof. Grune has previously studied the role of lipofuscin in proteasomal inhibition in human cell culture models using artificial lipofuscin. Later, he worked with isolated lipofuscin from human retinal epithelial cells and described the effects of this material on microglial cells. After securing a reliable source of human hearts, the Grune team began isolating real tissue lipofuscin. They are presently working to analyze composition and quantify degradation of LF. In recent years, the team has also worked with “artificial” lipofuscin and shown in a preliminary experiment that degradation by bacterial enzymes is possible. Upgrades to primary human material will allow optimization of the process of identifying bacterial enzymes with the ability to degrade the material.
Principal Investigator: Tilman Grune, PhD
Research Team: Annett Braune, Annika Höhn, Tim Baldesperger

As discussed in the project summary for “Glucosepane Crosslinks and Undoing Age-Related Tissue Damage”, adventitious crosslinking of collagen and elastin contributes to the slow stiffening of our arteries and other tissues with age. Some of these crosslinks occur as a result of spontaneous chemistry (as in the case of AGE crosslinking). Others, meanwhile, are the unintended consequences of metabolic processes that modify collagen — either as “collateral damage,” or to help us endure short-term problems at the cost of contributing to the long-term burden of crosslinking damage that eventually compromises function.
Amidst all of this, the sheer number of crosslinks of a given kind may not be a good measure of how high a priority it ought to be for rejuvenation biotechnology: some crosslinks may have a disproportionate effect on tissue elasticity depending on where they occur in the protein strand, how tightly they bind, and how much they interfere with the body’s ability break down and renew the tissue.
Recognizing the importance of prioritizing our targets, LRI is funding a study of this question at the Babraham Institute in Cambridge. In this study, “normally”-aging, nondiabetic mice are administered labeled building blocks for protein, which are then incorporated into extracellular matrix proteins, whose turnover can then be studied. The study has thus far provided valuable insight into how crosslinks regulate tissue stiffness, as well as updated our understanding of the turnover times of collagen and elastin. The results are currently under review and will be published soon.
Research Highlights:
One early and surprising finding of this work was that tissue crosslinks of types one might assume permanent are in fact continuously being broken apart and re-forming under the stress and strain of normal activity. It is the balance between these reversible crosslinks and the truly irreversible ones that gives rise to many of the changing mechanical properties of aging collagen.
The Babraham group have also confirmed that the crosslink profile in each tissue is distinct from others (which is only partially explained by the tissue-specific mixture of elastin and collagen), and that both the mixture of proteins and the pattern of protein-specific crosslinks changes with age. Importantly, some of the crosslinks that have been reported by others to accumulate in aging tissues were not detected. They are also complementing chemical analysis of the tissues with functional tests of the effect of these crosslinks on tissue mechanical function. The team have furthermore detailed chemical profiles of skin and tendon, and run pilot studies on a number of other tissues. Additional work is ongoing in mice fed with various sugars to further elucidate the role of sugars on collagen turnover in vivo.
Principal Investigator: Tilman Grune, PhD
Research Team: Annett Braune, Annika Höhn, Tim Baldesperger
Nearly half of the mammalian genome is long and short interspersed virus-like repetitive elements (LINEs and SINEs), which spread through the process of retrotransposition. Numerous intracellular mechanisms exist to silence these elements, but unfortunately these mechanisms decline with age. Active copies of these elements thus cause DNA damage over time as they integrate into genes and disrupt their functions. Cells with many such dysfunctions can mimic viral infections and trigger the production of the pro-inflammatory substance interferon (IFN). Normally, such cells are removed by the immune system, but as the immune system itself ages, it becomes less effective at this type of cleanup. This leads to “inflamm-aging”, which damages tissues further.
Analysis of blind mole rats, which exhibit both a natural IFN-driven mechanism for eradicating cells with damaged DNA as well as exceptionally long lifespans, has yielded observations supporting the LINE/SINE hypothesis.
LRI has sponsored a study to test the impact of the removal of damaged cells on healthspan and lifespan, via the creation of a special transgenic mouse at the Roswell Park Comprehensive Cancer Center.
Research Highlights:
This project began in the last quarter of 2019, and the Roswell Park team is now generating the transgenic mouse model. The group has already verified the transgene expression and activity in a cell culture model. Newborn mice are currently being screened for successful integration of the gene construct designed by the team. The final mouse model will allow the team to track the amount of IFN positive cells, and a lifespan study will begin later this year.

Principal Investigator: Andrei Gudkov, PhD, DSci
Research Team: Marina Antoch, Amy Stablewski, Nick Neznanov, Lilya Novototskaya, Olga Leontieva
Principal Investigator: Judith Campisi, PhD, Amit Sharma, PhD
Research Team: Abhijit Kale
Project Director: Julie Andersen, PhD
Research Team: Cyrene Arputhasamy, Manish Chamoli, Anand Rane

Project Director: Ruby Yanru Chen-Tsai, PhD
Principal Investigator: David Spiegel, PhD
Research Team: Prof. Jason Crawford, Nam Kim, Venkata Sabbasani, Matthew Streeter
Principal Investigator: Robert Shmookler-Reis
Principal Investigator: Jan Vijg
Research Team: Xiao Dong, Sylvia Gravina
Principal Investigator: Brian O’Nuallain
Research Team: Adam Cantlon
Principal Investigator: Daria Mochly-Rosen
Research Team: Gouri Yogalingam
Principal Investigator: Marisol Corral-Debrinski
Principal Investigators: Leonid Gavrilov, Natasha Gavrilova
Principal Investigator: Janko Nikolich-Zugich
Research Team: Megan Smithey
Principal Investigator: Irina Conboy
Research Team: Keith Causey, Justin Rebo
Principal Investigators: Barry Hughes, Randall Kuhn
Research Team: Eli Margolese-Malin, Dale Rothman, José Solórzano
