
Your #1 Source for Life Extension News
lifespan.io offers the latest information on rejuvenation biotechnology and life extension technologies. Our news outlet brings you the latest aging research, financial, and advocacy news, which is great if you like to keep up to date with everything happening in this rapidly changing field on a daily basis.
The Latest Longevity News Stories

Montana’s Right-to-Try Law Enters a New Phase
Montana’s first experimental treatment review board has brought three longevity heavyweights into the state’s effort to expand access to experimental therapies. How much regulation is too much? Many people […]
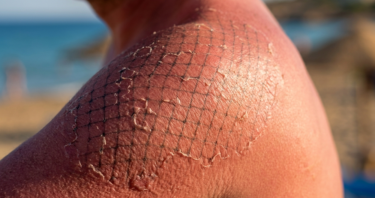
How the Immune System Makes Sun Damage Worse
In Aging Cell, researchers have published an explanation of how neutrophil extracellular traps (NETs) worsen UVB damage and how inhibiting them alleviated […]
NYC Woman Dead After Receiving a “Longevity Infusion”
According to the experts that we spoke to, the death, which followed an intravenous NAD⁺ infusion that went catastrophically wrong, underscores the risks of unproven treatments and the need for rigorous longevity medicine. Earlier this week, Elizabeth Baron, […]
More Autophagy Reduces Toxin-Induced Kidney Failure in Mice
Autophagy, which increases in younger mice under toxic stress, does not increase in older mice and leaves them susceptible to acute kidney injury (AKI). […]

The Progress and Future of Biological Aging Clocks
A recent review discusses the development of biological aging clocks, their capabilities and limitations, the discoveries they enabled, and possible future […]

Regular Training Erases Parts of Muscle Aging Signature
A new study suggests that regular planned exercise, not just being generally active, protects against certain aspects of muscle aging [1]. Are trained muscles younger? With age, skeletal muscles lose strength, metabolic flexibility, and mitochondrial capacity. While regular exercise slows […]

Gabriel Cian on Building the 2060 Longevity Ecosystem
In this follow-up interview, we speak with Gabriel Cian about how the 2060 ecosystem has evolved since last year’s Forum, what he learned from the first edition, and how he is […]
Exosomes From Stem Cells Fight Liver Disease in Mice
Researchers have described a method of using exosomes derived from mesenchymal stem cells (MSCs) to fight harmful metabolic changes in the liver. Another look at exosomes This is far from the first attempt at […]
Engineered Enzyme Reverses Age-Related Protein Damage
Scientists have engineered an enzyme that removes an advanced glycation end product (AGE) from proteins. This type of age-related modification, which affects protein function and can trigger inflammation, […]
Activating a Key Receptor Fights Thymic Involution in Mice
Publishing in Aging Cell, researchers have devised a way to delay the aging of the thymus by targeting GPR40, a key receptor in its epithelial cells. The organ that involutes The thymus trains a certain […]
Peripheral Inflammation May Drive Parkinson’s
A new study suggests that aging or Parkinson’s-triggering mutations create inflammation in peripheral tissues, and then circulating extracellular vesicles spread it to the brain, which might contribute […]
Combining Senolytics and Stem Cells Shows Promise in Mice
A new study associated with Immorta Bio suggests that combining a senolytic vaccine with mesenchymal stem cells might create a synergistic impact. However, the findings rest on […]
Interviewing the leading experts in aging research and longevity
We have a dedicated team of journalists who have interviewed many of the leaders in the field about their research and the drive to end age-related diseases. You can find our latest interviews below.




Regular Digest Articles
We publish the Rejuvenation Roundup – a monthly digest of what is happening in the field, a Longevity Market Recap – a monthly digest focused on the investment and business side of the field, and a quarterly Editorial – focusing on the activities of the news outlet and the wider organization.



Industry Press Releases
You can find some press releases from various companies in our field below. lifespan.io does not endorse any of these PRs and they are simply provided for information and interest.




Want even more news?
If you want to see even more recent articles, check out all news stories, or if you would like to look at a specific year or month head to the news archive.